



1. 初期理解
この段階では、先輩の前で間違いを避けるために、次のようないくつかの概念と用語を理解する必要があります。
Q: RT-PCR、qPCR、リアルタイム PCR、およびリアルタイム RT-PCR の違いは何ですか?
回答: RT-PCR は逆転写 PCR です。(逆転写 PCR、RT-PCR) は、ポリメラーゼ連鎖反応 (PCR) の変種として広く使用されています。RT-PCR では、RNA 鎖が相補的な DNA に逆転写され、それが PCR による DNA 増幅の鋳型として使用されます。
リアルタイム PCR および qPCR(定量的リアルタイム PCR) は同じもので、どちらもリアルタイム定量 PCR です。つまり、PCR の各サイクルにリアルタイムのデータ記録があるため、開始テンプレートの数を調整して正確な分析を行うことができます。
リアルタイム PCR (リアルタイム蛍光定量的 PCR) と逆転写 PCR (逆転写 PCR) はどちらも RT-PCR と略されるようですが、国際慣例では次のようになります。RT-PCR は特に逆転写を指します。PCR、リアルタイムPCRは一般にqPCR(定量的リアルタイムPCR)と略されます。.
リアルタイムRT-PCR(RT-qPCR)とは、逆転写PCRに蛍光定量技術を組み合わせたものです。: まず RNA 逆転写から cDNA (RT) を取得し、次にリアルタイム PCR を使用して定量分析 (qPCR) を行います。ほとんどの研究室は RT-qPCR、つまり RNA 発現のダウンレギュレーションに関する研究を行っているため、研究室で誰もが話す qPCR は実際には RT-qPCR を指しますが、臨床応用には依然として多くの DNA 検査があることを忘れないでください。B型肝炎ウイルスHBV検出などの定量分析。
質問: 蛍光定量 PCR を大量に読み取った後、増幅フラグメントを 80 ~ 300 bp の範囲内に制御する必要があるのはなぜですか?
答え: 各遺伝子配列の長さは異なり、いくつかは数 kb、いくつかは数百 bp ですが、プライマーを設計するときに必要なのは産物の長さが 80 ~ 300 bp であることだけであり、短すぎても長すぎても蛍光定量 PCR 検出には適していません。生成物のフラグメントは短すぎるため、プライマーダイマーと区別できません。プライマーダイマーの長さは30~40bp程度であり、80bp未満ではプライマーダイマーであるか生成物であるかを区別することが困難である。産物断片が300bpを超えて長すぎると、増幅効率が低下しやすく、遺伝子量を効果的に検出できなくなります。
たとえば、教室に何人いるかを数えるとき、口の数を数えるだけで済みます。遺伝子を検出する場合も同様で、遺伝子の特定の配列を検出するだけで十分です。配列全体で十分です。人を数えたい場合は、口と鼻、耳、眼鏡の両方を数える必要があり、間違いが起こりやすいです。
さらに拡大すると、生物学的研究では、点から領域まで多くの研究事例が存在します。どの種の遺伝子配列も非常に長いため、細菌の保存的配列を実行して特定の細菌集団の数を推測するための細菌 16S シークエンシングなど、すべての断片を測定することは不必要かつ不可能です。
Q: qPCR プライマー設計の最適な長さはどれくらいですか?
答え: 一般的に、プライマーの長さは約 20 ~ 24 bp であることがベターです。もちろん、プライマーを設計する際にはプライマーの TM 値に注意を払う必要があります。これは最適なアニーリング温度に関係するためです。多くの実験の結果、60°C がより良い TM 値であることが証明されました。アニーリング温度が低すぎると、非特異的な増幅が起こりやすくなります。アニーリング温度が高すぎると、増幅効率が相対的に低くなり、増幅曲線のピークが遅くなり、CT値が遅くなります。
Q: 色素法とプローブ法はどう違うのですか?
答え: 染色方法SYBR Green Ⅰ、PicoGreen、BEBO などの一部の蛍光色素は、それ自体では発光せず、二本鎖 DNA の副溝に結合すると蛍光を発します。したがって、PCR 反応の開始時には、機械は蛍光シグナルを検出できません。反応がアニーリング伸長段階に達すると、二本鎖が開き、DNA ポリメラーゼの作用により新しい鎖が合成され、蛍光分子が dsDNA 副溝に結合します。PCR サイクル数が増加するにつれて、より多くの色素が二本鎖 DNA と結合し、蛍光シグナルも継続的に強化されます。染色方法は主に科学研究に使用されます。
PS: 実験を行うときは注意してください。染料は人間の DNA と結合する必要があります。蛍光を発する人間に変わるように注意してください。
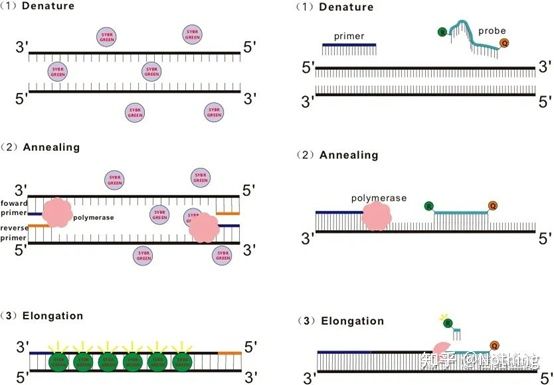
色素法(左) プローブ法(右)
PS: 実験を行うときは注意してください。染料は人間の DNA と結合する必要があります。蛍光を発する人間に変わるように注意してください。
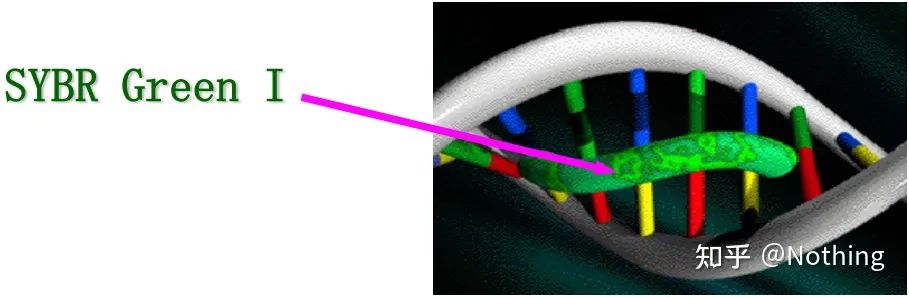
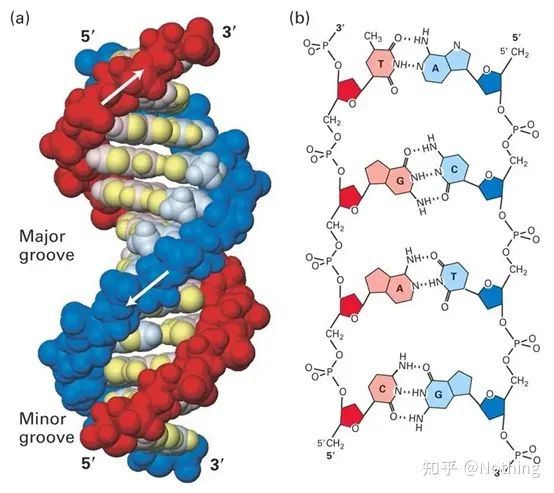
SYBR Green ⅠはDNAの副溝に結合します
プローブ法Taqman プローブは、最も一般的に使用される加水分解プローブです。プローブ(通常は FAM)の 5' 末端には蛍光基があり、プローブ自体は標的遺伝子に相補的な配列です。3'末端には蛍光消光基があります。蛍光共鳴エネルギー移動(フェルスター共鳴エネルギー移動、FRET)の原理によれば、レポーター蛍光基(ドナー蛍光分子)と消光蛍光基(アクセプター蛍光分子)が励起されると、スペクトルが重なり、距離が非常に近い(7〜10nm)場合、ドナー分子の励起によってアクセプター分子の蛍光が誘導される一方、自家蛍光は弱められます。したがって、PCR 反応の開始時に、プローブがシステム内で遊離しており無傷である場合、レポーター蛍光基は蛍光を発しません。アニーリングの際、プライマーとプローブはテンプレートに結合します。伸長段階では、ポリメラーゼは継続的に新しい鎖を合成します。DNA ポリメラーゼは 5'-3' エキソヌクレアーゼ活性を持っています。プローブに到達すると、DNA ポリメラーゼはテンプレートからプローブを加水分解し、レポーター蛍光基をクエンチャー蛍光基から分離し、蛍光シグナルを放出します。プローブとテンプレートは 1 対 1 の関係にあるため、検査の精度と感度の点でプローブ法の方が色素法よりも優れています。プローブ法は主に診断に使用されます。
Q: 絶対定量化とは何ですか?相対定量化とは何ですか?
答え: 絶対定量とは、血液 1 ml 中に HBV ウイルスが何個存在するかなど、qPCR で検査されるサンプルの初期コピー数の計算を指します。相対定量によって得られる結果は、別の参照サンプルに対する特定のサンプル中の標的遺伝子の量の変化であり、遺伝子発現は上方制御または下方制御されます。
Q: RNA 抽出量、逆転写効率、増幅効率は実験結果に影響しますか?
Q: サンプルの保存、抽出試薬、逆転写試薬、光透過性の消耗品は実験結果に影響しますか?
Q: 実験データを修正するにはどのような方法がありますか?
これらの問題については、以下の高度なセクションと高度なセクションで詳しく説明します。
2. 高度な知識
リアルタイム蛍光定量PCRに関しては、毎年数千件の科学研究論文が発表されており、その中には蛍光定量PCR技術も少なくないという現実を認識しなければなりません。
蛍光定量 PCR 実験を測定するための共通の標準がない場合、結果は大きく異なる可能性があります。同じ種の同じ遺伝子を同じ処理方法で使用した場合、検出結果も大きく異なり、後発者が同じ結果を繰り返すことは困難になります。あなたは、どれが正しくてどれが間違っているかは誰にもわかりません。
ということは、蛍光定量PCRはチート技術、あるいは信頼できない技術ということなのでしょうか?いいえ、蛍光定量 PCR の方が感度が高く、精度が高いため、操作を少し間違えると全く逆の結果が生じるからです。小さな損失は千マイルも離れたところにある。記事の著者は査読者から繰り返し拷問を受ける可能性があります。同時に、ジャーナルの査読者も、さまざまな実験結果から選ぶのが困難です。
総じて、リアルタイム PCR 実験におけるコンセンサスの欠如を指摘しています。この目的を達成するために、業界の上級科学者が標準を策定し始めました。これらの基準を満たすために、論文内で必要な実験およびデータ処理の詳細 (必要なデータを含む) を提供することを寄稿者に要求します。
レビュー担当者は、これらの詳細を読むことで実験の品質を判断できます。今後の読者もこれを使用して実験を繰り返したり、実験を改善したりすることができます。こうして得られた実験結果は、情報量が多く、質が高く、使えるものになります。
MIBBI (生物学的および生物医学的調査のための最小限の情報 -http://www.mibbi.org)が誕生しました。MIBBI は実験の標準を提供するプロジェクトです。自然界に公開されています。このプロジェクトは、細胞生物学、マイクロアレイ、これから説明するqPCRなどを含むさまざまな生物学的実験を対象としており、原稿を投稿する際に実験の種類ごとに規定されています。その情報は常に提供されるべきです。
MIBBI プロジェクトには、蛍光定量 PCR に関連する 2 つの論文があります。:
·RDML (リアルタイム PCR データ マークアップ言語) – リアルタイム定量 PCR データの構造化言語およびレポート ガイド。
·MIQE (定量的リアルタイム PCR 実験の出版のための最小限の情報) – リアルタイム定量的 PCR 実験に関する論文を出版するための最小限の情報。
まず、用語仕様である RDML について説明します。
すべてに標準的な定義がなければ議論を続けることは不可能であるため、試験では用語の説明が非常に重要です。
蛍光定量PCR実験で使用される用語には以下の内容が含まれます。QIAGEN が私たちに最適な要約を作成してくれました。以下はすべてドライです品 .
増幅曲線
増幅曲線は、サイクル数を横軸、反応中のリアルタイム蛍光強度を縦軸として、PCR プロセス中に作成された曲線を指します。
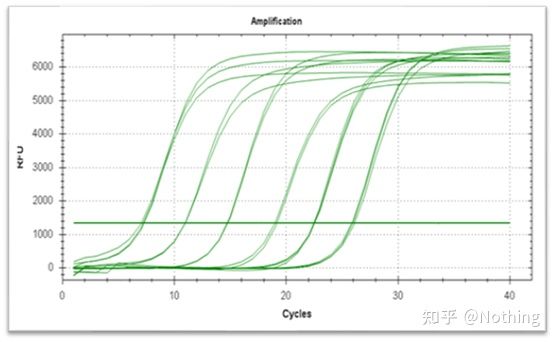
優れた増幅曲線は次の特性を備えている必要があります。: ベースラインは横ばいまたはわずかに減少しており、明らかな上昇傾向はありません。曲線の変曲点は明確であり、指数関数的位相の傾きは増幅効率に比例します。傾きが大きいほど、増幅効率は高くなります。全体的な増幅曲線 平行度は良好で、各チューブの増幅効率が類似していることを示しています。低濃度サンプルの増幅曲線の指数関数的段階は明らかです。
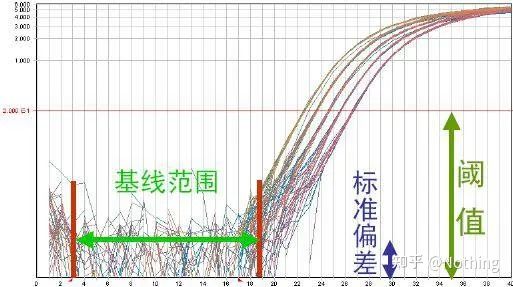
ベースライン (ベースライン)
ベースラインは初期サイクルのノイズ レベルです。、通常、3 サイクル目と 15 サイクル目の間で測定されます。これは、この期間では増幅産物によって引き起こされる蛍光値の増加を検出できないためです。ベースラインの計算に使用されるサイクル数は変動する可能性があり、多量のテンプレートを使用する場合、または標的遺伝子の発現レベルが高い場合には、サイクル数を減らす必要がある場合があります。
ベースラインを設定するには、直線性増幅曲線から蛍光データを確認する必要があります。ベースラインは、増幅曲線の成長がベースラインのサイクルトップ番号より大きいサイクル番号で始まるように設定されます。ベースラインはターゲット配列ごとに個別に設定する必要があります。初期のサイクルで検出された平均蛍光値は、増幅産物で得られた蛍光値から差し引く必要があります。さまざまなリアルタイム PCR ソフトウェアの最新バージョンでは、個々のサンプルのベースライン設定を自動的に最適化できます。
PCR 増幅反応の最初の数サイクルでは、蛍光シグナルはあまり変化しません。直線に近づくことをベースラインと呼びますが、最初の数サイクルをよく見ると、ベースライン内で下の図で何が起こっていることがわかります。
背景 背景とは、
反応における非特異的蛍光値。例: 非効率的な蛍光消光。またはSYBR Greenの使用による多数の二本鎖DNAテンプレート。シグナルのバックグラウンド成分は、リアルタイム PCR ソフトウェア アルゴリズムによって数学的に除去されます。
レポーター信号
レポーターシグナルとは、リアルタイム PCR 中に SYBR Green または蛍光標識された配列特異的プローブによって生成される蛍光シグナルを指します。
正規化されたレポーター信号 (RN)
RN は、各サイクルで測定されたレポーター色素の蛍光強度をパッシブリファレンス色素の蛍光強度で割ったものを指します。
パッシブリファレンス色素
一部のリアルタイム PCR では、蛍光色素 ROX は、蛍光シグナルを正規化するための内部基準として使用されます。。不正確なピペッティング、ウェルの位置、蛍光の変動による変動をウェルごとに補正します。

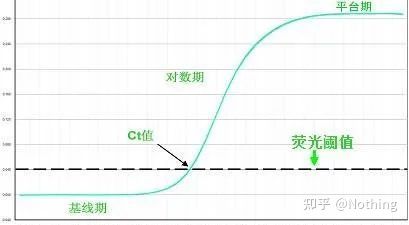
蛍光閾値(しきい値)
増幅曲線のバックグラウンド値よりも高く、プラトー値よりも大幅に低く調整されました。これは、PCR 検出の対数直線範囲を表す増幅曲線の直線領域内になければなりません。PCR の対数直線相を簡単に識別できるように、対数増幅曲線ビューでしきい値を設定する必要があります。リアルタイム PCR ではターゲット遺伝子が複数ある場合、ターゲットごとに閾値を設定する必要があります。一般に、PCR 反応の最初の 15 サイクルの蛍光シグナルが蛍光バックグラウンドシグナルとして使用され、蛍光閾値は PCR の最初の 3 ~ 15 サイクルの蛍光シグナルの標準偏差の 10 倍であり、蛍光閾値は PCR 増幅の指数関数期に設定されます。一般に、各機器には使用前に蛍光閾値が設定されています。
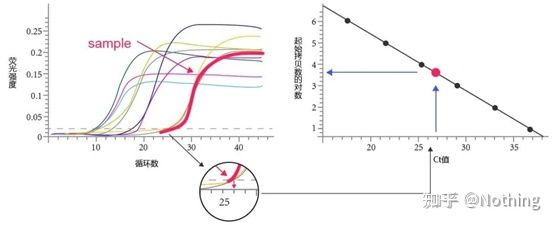
サイクルしきい値 (CT) またはクロッシングポイント (CP)
増幅曲線が閾値と交差するサイクル (つまり、蛍光検出が大幅に増加する点)。CT は分数にすることができ、開始テンプレートの量を計算できます。CT 値は、各 PCR 反応チューブ内の蛍光シグナルが設定された閾値に達するときのサイクル数を表します。各テンプレートの CT 値とテンプレートの初期コピー数の対数の間には線形関係があります。初期コピー数が大きいほど CT 値は小さくなり、その逆も同様です。標準曲線は、既知の初期コピー数を有する標準を使用することによって作成することができ、横軸はCT値を表し、縦軸は初期コピー数の対数を表す。したがって、未知サンプルのCT値が得られれば、標準曲線からサンプルの初期コピー数を計算することができる。
ΔCT値
ΔCT値は次のとおりです標的遺伝子と対応する内因性参照遺伝子のCT値の差、ハウスキーピング遺伝子など、使用されるテンプレートの量を正規化するために使用されます。
⇒ΔCT = CT (標的遺伝子) – CT (内因性参照遺伝子)
ΔΔCT値
ΔΔCT値は、対象サンプル(例えば、刺激された細胞)の平均ΔΔCT値と参照サンプル(例えば、刺激されていない細胞)の平均ΔΔCT値との間の差を表す。参照サンプルはキャリブレーション サンプルとも呼ばれ、他のすべてのサンプルは相対的な定量化のためにこれに正規化されます。
⇒ΔΔCT = 平均ΔCT (対象サンプル) – 平均ΔCT (参照サンプル)
内因性参照遺伝子 (内因性参照遺伝子)
ハウスキーピング遺伝子(ハウスキーピング遺伝子)などの内因性参照遺伝子の発現レベルはサンプル間で異なりません。参照遺伝子の CT 値をターゲット遺伝子と比較すると、ターゲット遺伝子の発現レベルを入力 RNA または cDNA の量に対して正規化できます (上記の ΔCT 値のセクションを参照)。
内部参照遺伝子が正しいRNA 分解の可能性や RNA サンプル中の酵素阻害剤の存在、さらには RNA 含有量、逆転写効率、核酸回収、サンプル取り扱いの変動などです。最適な参照遺伝子を選択するために、実験設定に応じて最適な参照遺伝子を選択できるようにアルゴリズムを変更しました。
内部制御
ターゲット配列と同じ反応で増幅され、異なるプローブでプローブされる (つまり、デュプレックス PCR を実行する) コントロール配列。内部コントロールは、標的配列が検出されない場合など、増幅の失敗を除外するためによく使用されます。
校正サンプル
遺伝子の相対発現レベルを決定するために他のすべてのサンプルを比較するための相対定量に使用される参照サンプル (細胞株または組織からの精製 RNA など)。キャリブレーション サンプルは任意のサンプルにすることができますが、通常は対照 (未処理のサンプルや実験のゼロ時点からのサンプルなど) です。
ポジティブコントロール
制御反応を使用して既知の量のテンプレート。ポジティブコントロールは、プライマー セットまたはプライマー - プローブ セットが適切に機能していること、および反応が正しく設定されていることを確認するためによく使用されます。
テンプレート制御なし (NTC)
テンプレートを除く増幅反応に必要なすべての成分を含むコントロール反応。通常は水に置き換えられます。NTC を使用すると、試薬の汚染や外来 DNA による汚染を検出できるため、検出データの信頼性と信頼性が保証されます。NTC コントロールの増幅は汚染を示します。
RT制御なし(NRT)
RNA 抽出プロセスには残留ゲノム DNA が含まれる可能性があり、これは非常に有害であり、データ品質に影響を与える元凶であり、qPCR の天敵であるため、実験を設計する際には、RNA 検出のみを増幅するように設計する必要があります。方法には 2 つあり、1 つはイントロンをまたぐプライマーを設計する方法、もう 1 つは DNA を完全に除去する方法で、どちらが優れているかについては後述します。NTRコントロールはDNA汚染を検知する魔法の鏡。増幅がある場合、それは汚染があることを意味します。
規格
標準とは、標準曲線を作成するために使用される既知の濃度またはコピー数のサンプルです。標準の安定性を確保するために、通常、遺伝子断片をプラスミドにクローン化し、標準として使用します。
標準曲線
通常、標準品を用いて倍増比に応じて少なくとも5つの濃度勾配に希釈し、CT値とコピー数の座標上に5つの点を描き、それらの点を直線状に結んで検量線を作成します。各標準曲線について、その有効性をチェックする必要があります。傾きの値は -3.3 ~ -3.8 の間にあり、各濃度は 3 回実行されます。他の点と著しく異なる点は破棄する必要があります。被検試料のCT値を標準曲線に当てはめることにより、被検試料の発現量を算出することができる。
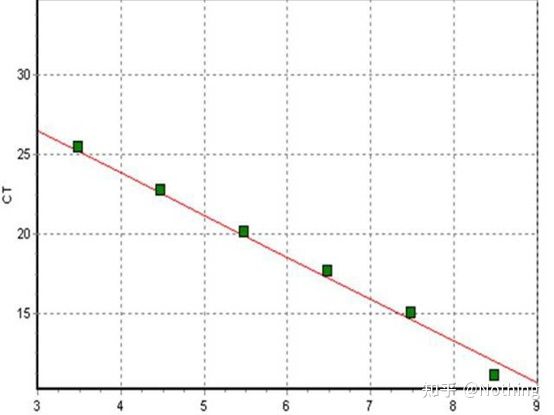
検査対象サンプルのCT値を標準曲線に取り込み、検査対象サンプルの初期コピー数を計算することができます。
効率と傾き
標準曲線の傾きは、リアルタイム PCR の効率を表します。
· -3.322 の傾きは、PCR 増幅効率が 1、つまり 100% 効率であり、PCR 産物の量が各サイクルで 2 倍になることを示します。
· –3.322 未満の傾き (例 –3.8) は PCR 効率を示します。
· –3.322 (例: –3.0) より大きい傾きは、PCR 効率が 100% を超えているように見えることを示します。これは興味深いことですが、PCR の 1 サイクルで 2 倍を超える増幅産物がどのように生成されるのでしょうか?この状況は、PCR 反応の非線形段階で発生します。つまり、大量の非特異的増幅が発生します。
融解曲線
qPCR 増幅が完了したら、PCR 産物を加熱します。温度が上昇すると二本鎖増幅産物が徐々に融解し、蛍光強度が低下します。特定の温度 (Tm) に達すると、多くの製品が溶けます。蛍光が急激に低下します。異なる PCR 製品は異なる Tm 値と異なる融解温度を有するため、PCR の特異性を識別できます。
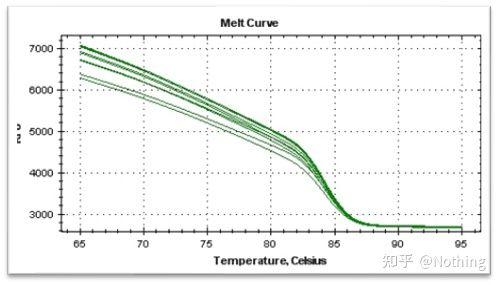
融解曲線(微分曲線)
融解曲線を導出してピークマップを作成することで、PCR産物フラグメントの状況をより直感的に表示できます。融解温度は DNA 断片の Tm 値であるため、断片サイズ、GC 含有量など、DNA 断片の Tm 値に影響を与えるいくつかのパラメーターを判断できます。一般的に、当社のプライマー設計原則によれば、増幅産物の長さは 80 ~ 300 bp の範囲にあるため、融解温度は 80°C ~ 90°C にする必要があります。
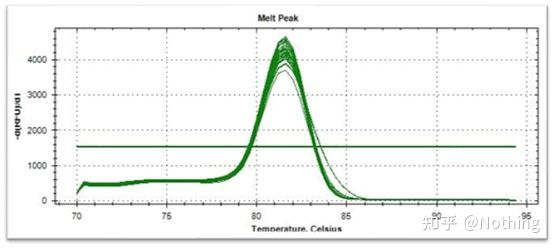
融解曲線の解釈: 80°C ~ 90°C の間にメインピークのみが現れる場合は、蛍光定量 PCR が完全であることを意味します。メインピークが 80°C ~ 90°C の間に現れ、その他のピークが 80°C 未満に現れる場合、基本的にプライマーダイマーが考慮されます。この問題を解決するには、アニーリング温度を上げることを試みることができます。80℃~90℃でメインピークが現れ、温度が上がると再び雑ピークが現れる場合は、基本的にはDNA汚染があると考えられ、実験の初期段階でDNAを除去する必要があります。
もちろん、まだ異常な状況はいくつかあります。以下でそれを 1 つずつ説明します。
3. 高度な知識
qPCR を行うには、MIQE と言わなければなりません。最低限の情報の出版のために定量的リアルタイム PCR実験 - リアルタイム定量 PCR に関する論文を出版するための最小限の情報実験。皆様にわかりやすくするために、重要な内容を簡略化して記載させていただきます。
MIQE の原文はインターネットで検索できますが、最も重要なことは、次のことが規定されていることです。記事を公開するときに提供する必要があるデータチェックリスト .
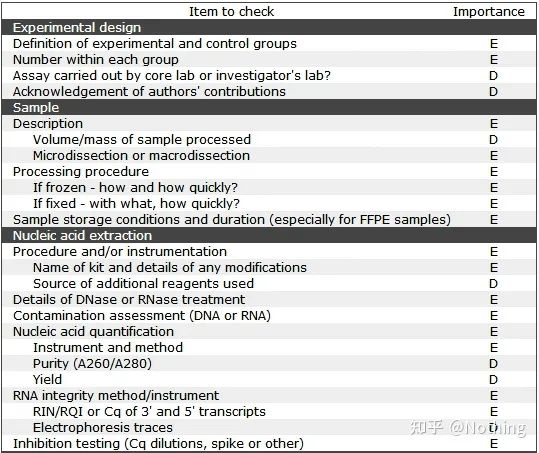
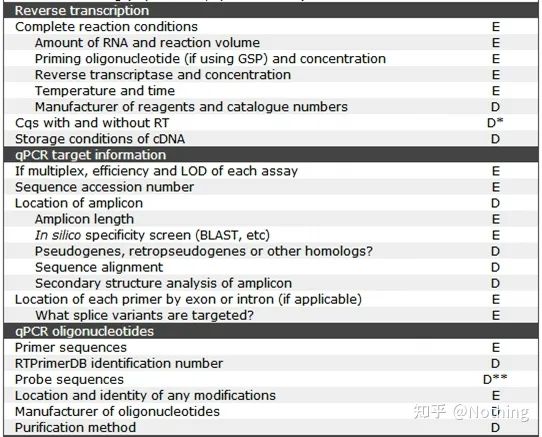
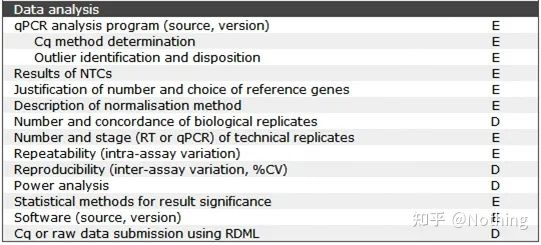
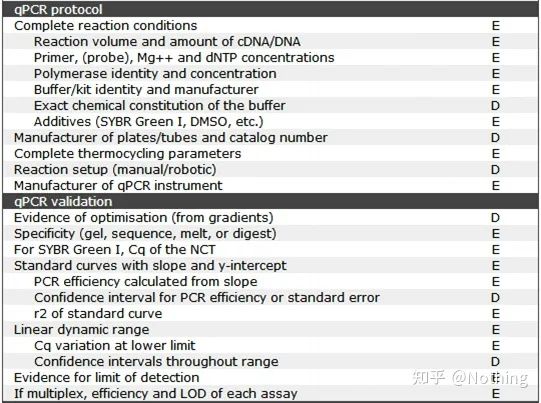
レビュー担当者は、これらの詳細を読むことで実験の品質を判断できます。今後の読者もこれを使用して実験を繰り返したり、改良したりすることができます。
このリストでは、各リストの重要性がそれぞれ E または D でマークされていることは注目に値します。どういう意味ですか?E: 必須情報 (必ず提出してください);D: 望ましい情報(できるだけ多くの情報を提供してください)。
MIQE (1) - 実験デザイン
大学院を卒業して弁護を終えた多くのクズ野郎は、自分で実験を計画し、ノートを開いて、教師の指示に従って行動する方法を知らないでしょう。結果的に実験設計は厳密なものではなく、雑誌編集部が「この絵とあの絵を作りたい」と言いながら、ボーッとやりました。クソ野郎はこうして作られるんだ!

もっと身近なところで言えば、実験の第一原則は次のことを決定することです。実験ロジックの厳密さ。最も基本となるのは実験計画法であり、実験計画法で最も重要なのは、実験データを参照し、比較し、納得させるために、対象サンプル、参照サンプル(コントロール)、繰り返し回数をどのように設定するかです。
対象サンプル特定の処理後に標的遺伝子を検出する必要があるサンプルを指します。参考サンプル何も処理されていないサンプルであり、生物学では野生型と呼ばれることがよくあります。
実験の反復はとても重要です。一般に、説得力のある反復の数は 3 つ以上である必要があります。何が生物学的複製であり、何が技術的複製であるかを区別する必要がある。
生物学的複製: 異なる材料 (時間、プラント、バッチ、反応プレート) を使用して行われた同じ検証実験。

生物学的重複
コショウの農薬処理を例に挙げてみましょう。ABC の 3 つの植物に農薬を散布したいとします。すると、ABC の 3 つの植物は 3 つの生物学的複製であり、異なる材料を使用して行われた同じ検証実験です。しかし、実験としては必ず対照が必要なので、植物 A の枝の 1 つに噴霧して植物 A の実験グループを形成し、植物 A の他の枝には噴霧しないで対照グループを形成することができます。BとCについても同じことを行います。
テクニカルレプリケート (テクニカルレプリケート):操作による誤差を避けるために繰り返された実験であり、実際には同じ材料に含まれる重複した穴です。処理とコントロールの両方に、標的遺伝子と内部参照遺伝子の反復設定 (最低 3 つ) が必要です。
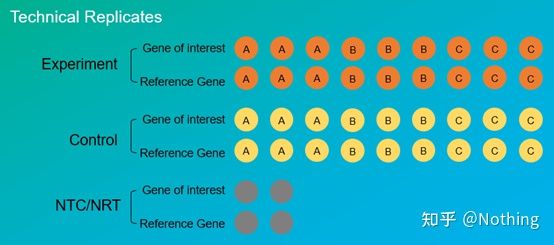
技術的な繰り返し
もう一度農薬で処理されたピーマンを例に挙げてみましょう。植物Aの実験群については、標的遺伝子と内部参照遺伝子に対してそれぞれ1、2、3のPCRホールを3つ開け、検出後の平均値をとりました。植物 A の防除についても同様に処理します。同様に、B と C の植物にも同じ処理を行います。これは技術的な繰り返しです。
注目に値するのは、統計に入るのは生物学的な反復であり、技術的な反復は、実験結果の信頼性を高めるために、実験プロセスにランダムな現象が存在するかどうかをテストすることです。つまり、よく言われるように、平均を取ることでエラーを回避します。
ネガティブコントロール - NTC および NRT
NTC (テンプレートなし制御)、テンプレートのないコントロールは、実験材料が汚染されているかどうかを確認するために使用されます。一般に、水がテンプレートとして使用されます。蛍光反応がある場合は、実験室で核酸汚染が発生したことを示します。
これらの汚染は、不純な水、内在性 DNA を含む不適格な試薬、プライマー汚染、実験室設備汚染、エアロゾル汚染などによって引き起こされ、RNase スカベンジャーと RNase 阻害剤を使用する必要があります。エアロゾル汚染は発見するのが最も困難です。あなたの研究室が空気中にさまざまな核酸が浮遊しているスモッグのようなものだと想像してください。

NRT (非逆転写酵素)逆転写のないコントロールは、ネガティブコントロールとしての逆転写されていないRNAであり、gDNA残基のコントロールです。
遺伝子発現を行う場合、逆転写後のcDNA量を検出することでRNA量を検出します。実際に得られる結果は gDNA と cDNA であるため、RNA 精製時に gDNA 残基が存在すると実験結果に誤差が生じます。cDNA だけでなく、凝集体レベルでも、RNA 抽出中に gDNA を完全に除去する必要があります。
MIQE (2) - サンプル情報
いわゆるサンプル情報ということは、qPCRに関する論文を出版する場合には、サンプル情報をわかりやすく説明する必要があり、これは論文の必須の部分です。同様に、サンプルを処理する場合も、サンプルの有効性を確保するために独自の操作を規制する必要があります。

サンプルの説明は単なる結果であり、実験全体で採取された材料にはもっと注意を払う必要があります。
実験材料の選択
血液サンプル – 4 時間以内の新鮮な血液を選択してください。細胞サンプル – 活発な増殖期に新鮮な細胞を収集することを選択します。動物組織 - 新鮮で活発に成長している組織を選択してください。植物組織 – 新鮮な若い組織を選択してください。
これらのいくつかの文の中に、「 fresh 」というキーワードがあることに気付いたはずです。
上記のサンプルの場合、市場で最もコスト効率が高く安定したキットは、DNA と RNA を迅速かつ簡単に抽出できる Foregene のキットです。
実験材料の保管
一般に、条件が許せばサンプルを保管することはお勧めしません。しかし、サンプリング後すぐに実験ができない友人も多く、サンプリングのために液体窒素タンクを現場まで運ぶ必要がある人もいます。
このような勤勉な友人にしては、試薬の消耗品について理解していないとしか言いようがありません。現在、多くの試薬消耗品会社が RNA サンプルを室温で保存できる試薬を製造しており、それらを選択して使用できます。従来の保管方法は、持ち運びが容易な小型の液体窒素タンクを使用した液体窒素保管です。サンプルを研究室に持ち帰った後は、-80°C の冷蔵庫に保管してください。

RNA を含む実験では、6 ワードの原則に従う必要があります。低温、酵素なし、と速い .
低温の概念は理解しやすいです。酵素がなければ、RNase は私たちが住んでいる世界のどこにでも存在します (そうでなければ、HIV によって殺されていたでしょう)。そのため、実験を行うときに RNase をどのように回避するかは非常に重要な概念です。速い、この世に破られないカンフーはない、破られないのはスピードだけだ。
したがって、ある意味、抽出時間が短いほど良いキットであると言えます。どしてフォアジーンのキットはスピードを重視しています。彼らはそれをよく知っているからです。
追伸: 非常に慎重に実験を行う女の子もいますが、数年間の努力を経て完成するほどの成果は得られません。彼らは、神は不公平で、他人について不平を言い、命を求めていると感じています。実際、彼女はそれを理解していませんでした。彼はRNAをうまく守れず、スラムダンク選手は機敏でした。実験中は3回、5回、2回でスラムダンクを終わらせるだろうと思っていたそうですが、見事に実験を行うことができました。
ノート: RNase 侵入の速度が遅くなり、可能性が高くなります。速くなるためのトレーニング方法は?仕方ない、もっと練習するしかない。
異なる実験や異なるサンプルについては、より多くの文献を読み、適切な処理方法を選択する必要があります。MIQE では、サンプルの収集と保管のプロセスについて、論文に明確に記載することを義務付けています。これにより、査読者が論文の信頼性をレビューできるようになります。また、唖然とした若者が実験を繰り返すのにも便利です。
生物実験は難しいですが、ハイエンドです。気をつけないと世界をひっくり返す可能性があります。たとえば、SARSを生化学的危機にするとか、ハイブリッド米を作って13億人を救うとか。下の写真は化学実験の様子ですが、そのチンコのような姿を見ただけでも、自分の研究にどれだけ誇りを持っているかが分かるはずです。忘れてください、彼を黒くしないでください。

MIQE (3) – 核酸抽出。
核酸抽出は一大イベントであり、分子生物学の実験はすべて核酸抽出から始まります。まずは、MIQEの核酸抽出に関する内容をコピーしてみましょう。
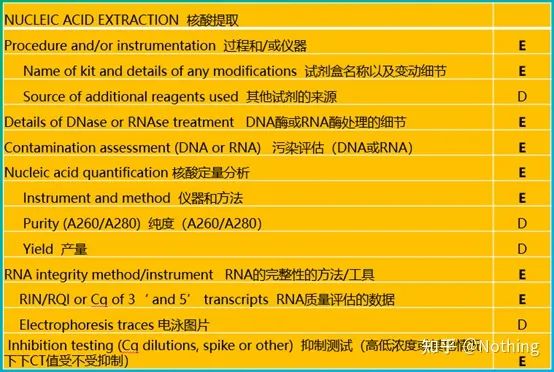
この姿を見ていると、表面に留まっているわけにはいきません。形式は定説です。優秀な学生になるには、その理由を尋ねる必要があります。この表の重要な内容は次のとおりです。RNAの純度、完全性、一貫性、抽出量 .
の最初の部分プロセスまたは機器は核酸抽出ステップです。自動核酸抽出装置を使用して抽出する場合(上級者向け、ご購入の際はご相談ください)、装置のモデル名を指定する必要があります。

キットの名前と
他の人があなたの実験を簡単に再現できるように、変更の詳細にどのキットが使用されたか、どのような特別な試薬が追加されたか、またはどのような特別な操作が行われたかを明確に説明する必要があります。
特別なサンプルを抽出するときに、これを秘密兵器だと思って特別な試薬を追加し、他の人には言わない人もいます。それを秘密にしている間、彼らはあなたの記事を輝かせる機会も失います。賢くならないでください、科学研究において田舎の張老人よりも正直でなければなりません、もし賢くなりたければ、記事はあなたを愚かにするでしょう。
キットの製品番号を覚えておく必要がありますキットを注文して記事を書くとき。通常、キットには 2 つの番号があります: Cat - カタログ番号 (製品番号、品目番号)、Lot - 製品ロット番号 (製品がどのバッチから来たかを示すために使用されます)。
また、CAS番号は生化学試薬を注文する際によく使われますので、併せて普及させていきます。CAS 番号は、米国化学会によって各新しい化学薬品に与えられる番号です。通常、3 つの数字はダッシュで接続されます。Rushui の CAS 番号: 7732-18-5。化学物質には複数の別名があることがよくありますが、CAS 番号は一意です。医薬品を注文するときは、まず CAS 番号を確認します。
もっと身近な話ですが、なぜこれらのことを明確に説明する必要があるのでしょうか?実は、RNA抽出の品質をチェックするためでもあります。機器やキットを使用すると、RNA 抽出がより安定します。通常の研究室での抽出規模は大きくなく、キットで入手可能です。
DNaseまたはRNase処理の詳細
蛍光定量 PCR の重要な問題は、DNA の汚染を防ぐことであり、汚染がある場合は実験を行わないことです。したがって、実験過程で DNA が完全かつ完全に除去されたことを証明するために、DNA の処理に使用したプロセスを記載することが不可欠です。模式図で表します。
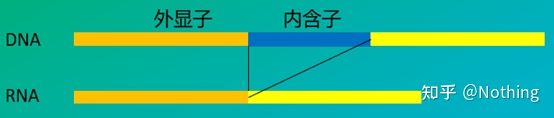
RNAとDNAの模式図
DNAを除去する方法としては、RNAを抽出した後にDNaseで処理するのが一般的です。ただし、これらは比較的古い方法です。市販の RNA 抽出キットは、抽出プロセス中に DNase を添加せずに DNA を除去できます。たとえば、Foregene の一連のキットです。
ノート: RNA 抽出中に DNA を除去することは非常に危険な諸刃の剣であり、RNA 抽出の操作時間が延長され、RNA 分解のリスクが高まります。基本的に、RNA 収量と純度はトレードオフになります。
また、シリカ系吸着カラムに添加するDNaseの量は非常に少量であるため、効果を得るには高品質のDNaseを使用する必要があります。最適化されていない DNase は、迅速かつ完全に消化できません。これは販売者の技術レベルをテストするものです。もちろん、DNase を使わずに DNA を除去できると豪語するさらに奇妙な業者もいます。DNase を使わずに DNA を完全に除去できると豪語する人はフーリガンであると言えます。DNAは比較的安定した二本鎖構造であり、話したり笑ったりしただけでは消去できません。
汚染評価
評価方法: 電気泳動検出、1% アガロース、6V/cm、15 分、負荷 1-3 ul
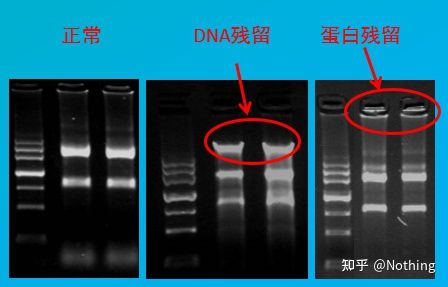
核酸定量分析
通常、UV分光光度計を使用して測定されます。まず、OD260、OD280、OD230 の 3 つの値の意味を説明します。
・OD260nm:核酸の最も高い吸収ピークの吸収波長であり、最良の測定値は0.1~1.0の範囲となります。そうでない場合は、サンプルを希釈または濃縮して範囲内にします。
・OD280nm:タンパク質やフェノール性物質の最大吸収ピークの吸収波長です。
・OD230nm:炭水化物の最も高い吸収ピークの吸収波長です。
次に、各指標の役割について説明します。A260 の場合、核酸の収量の測定に使用できます。OD260=1、dsDNA=50μg/ml、ssDNA=37μg/ml、RNA=40μg/mlの場合。
純度については、一般的に見られる比 (OD260/280 および OD260/230) に注目する必要があります。
・純粋なDNA:OD260/280は約1.8に相当します。1.9 より大きい場合は RNA 汚染があることを示し、1.6 より小さい場合はタンパク質とフェノール汚染があることを示します。
・純粋RNA:1.7
・OD260/230:DNA、RNAを問わず基準値は2.5です。2.0未満の場合、砂糖、塩分、有機物の汚染があることを示します。
RNAの完全性
RNA の完全性を測定することは非常に重要です。一般に、28S RNA と 18S RNA の明るさが 2 倍の関係にあるかどうかを確認するには、RNA 変性ゲル実験を行う必要があります。3 番目のバンド 5S が表示されると、無脊椎動物を除いて RNA が分解し始めたことを意味します。

RNA 品質評価用のデータ: 上記のテストに加えて、RNA の完全性に関しては、目に見えない形で RNA が分解されているかどうかを検出できる Experion 自動電気泳動システムの RQI 完全性テストなど、より高度な機器テストもいくつかあります。
科学研究における蛍光定量 PCR は、標的遺伝子と内部参照遺伝子の比較です。したがって、RNA サンプルの保存、RNA 抽出などのプロセスにおける主な目標は、RNA の完全性を確保することです。
RNA の完全性がターゲット遺伝子と内部参照遺伝子の間のバランスにどのような影響を与えるかは、以下の図から簡単に理解できます。分解は遺伝子の不完全性につながり、それが内部参照遺伝子の不完全さであっても、ターゲット遺伝子の不完全さであっても、データに大きな影響を与えます。

ターゲット遺伝子とリファレンス遺伝子の模式図は、真実であってはなりません
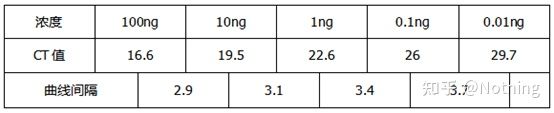
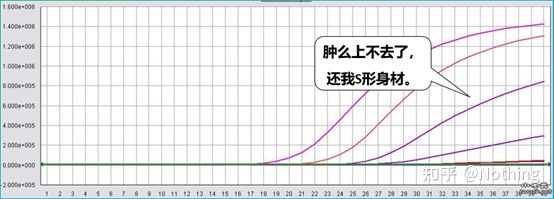
阻害試験(高濃度、低濃度等の条件下でCT値が抑制されるかどうか)
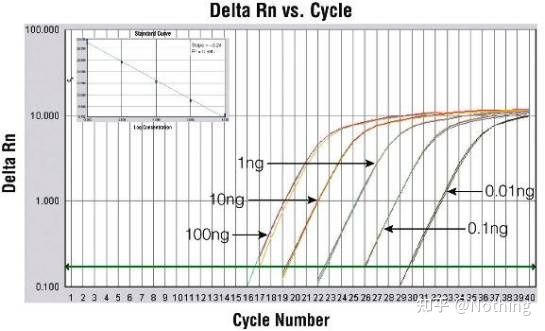
この図を例にとると、5 つの曲線の Ct 値は次のようになります。曲線間の CT 値の分布は不均一であり、Ct 値は高濃度および低濃度下で遅延します。これは PCR 阻害の場合です。
重要なポイント: RNA 抽出のプロセスでは、誤解を捨てて正しい概念を確立する必要があります。
間違った考えは、RNA 抽出では収率のみを追求し、得られる RNA の量が多ければ多いほど良いと考えることです。実際、定量を行う場合、遺伝子の数がそれほど多くなければ、RNA はそれほど必要ありません。抽出する RNA の量は十分すぎるほどです。
正しい概念は次のとおりです。RNA抽出では純度、完全性、一貫性を追求する必要があります。純度が高いと、その後の逆転写が阻害されず、データが DNA の影響を受けないことが保証されます。完全性により、ターゲット配列と内部参照のバランスが確保されます。一貫性により、安定したサンプルローディングが保証されます。
MIQE (4) – 逆転写
誤解:より高いサンプル量の追求。
正しい概念:一貫性(安定性)を追求し、RNA量に関わらず逆転写効率が安定し、cDNAの違いがmRNAの違いを忠実に反映します。
このプロセスを概略図で説明します。
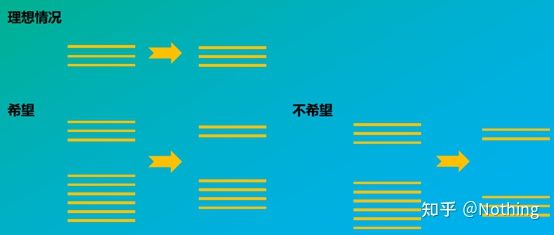
逆転写効率の模式図、真実ではない
まず最初に、逆転写プロセスと PCR プロセスの違いを理解する必要があります。PCR では複数回の加熱とアニーリングのプロセスが行われ、ターゲットのフラグメントは指数関数的に増加します。逆転写にはこのプロセスはありませんが、逆転写は実際には 1 対 1 で行われると想像できます。複製プロセスでは、RNA の数と同じ数だけ
cDNA 情報はいくらでも取得できますが、大小さまざまな断片が逆転写されており、1 つの断片に焦点を当てることは不可能であるため、もう理解されているはずです。また、増幅効果のあるPCRとは異なり、RNAの量が比較的少ないため、得られるcDNAの量も比較的少なく、基本的に検出することはできません。
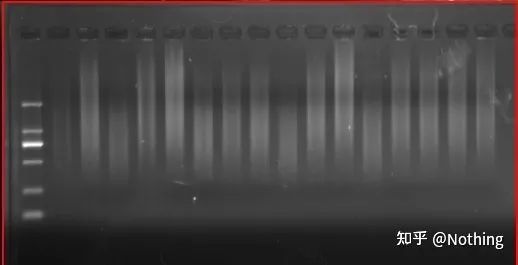
cDNA電気泳動結果
第二に、理想的には逆転写は 1 対 1 で実行されますが、どの企業の逆転写酵素もこの効果を達成することはできません。基本的に、ほとんどの逆転写酵素の効率は 30 ~ 50% の間で変動します。この場合、比較的安定した逆転写効率が得られると考えられます。これを図で確認します。3 つの RNA は 2 つの cDNA を取得し、6 つの RNA は 4 つの cDNA を取得するため、ロードされるサンプルの量に関係なく、逆転写効率は比較的安定しています。逆転写効率が不安定で高濃度が阻害される事態は見たくない。
では、逆転写効率が安定しているかどうかを確認するにはどうすればよいでしょうか?方法は非常に簡単で、比較試験を行うだけで済みます。1 つは RNA を 2 倍希釈した後に cDNA に逆転写し、もう 1 つは cDNA に逆転写した後に 2 倍希釈し、その後 qPCR を行って得られた傾きを確認することです。これは一致していますか。優等生であれば、数秒で理解できるはずです。以下に示すように:
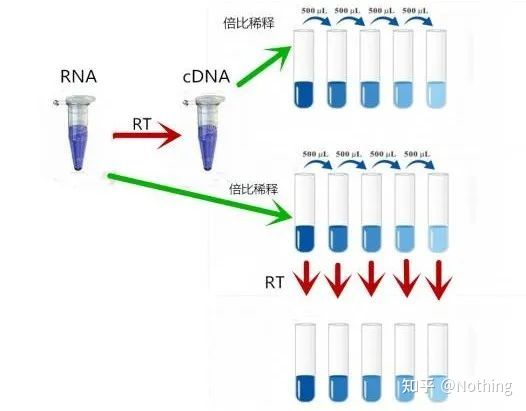
RNA と cDNA を希釈して逆転写効率が安定しているかどうかをテストします。
逆転写酵素とキット
完璧な蛍光定量 PCR には優れた逆転写酵素とキットが必要です。逆転写酵素は発生源によりAMVとAMVの2種類に大別されます。M-MLVであり、その性能は表に示すものと同じです。
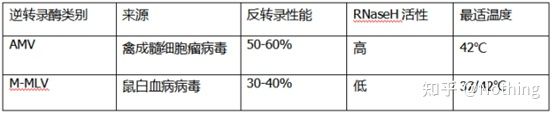
RNase H活性
RNase HはリボヌクレアーゼH、中国名はリボヌクレアーゼHで、DNA-RNAハイブリッド鎖のRNAを特異的に加水分解できるエンドリボヌクレアーゼです。RNase H は、一本鎖または二本鎖の DNA または RNA のホスホジエステル結合を加水分解できません。つまり、一本鎖または二本鎖の DNA または RNA を消化できません。cDNA の 2 番目の鎖の合成に一般的に使用されます。
不思議なことだ。逆転写酵素が RNase H 活性を持っていると言っているのは、逆転写酵素に RNase H が含まれているということではありません。また、おそらく逆転写酵素の特定のグループの構造が原因で、逆転写酵素から RNase H を分離することができない可能性があります。この活性は逆転写酵素によって引き起こされます。
したがって、AMV の逆転写効率が高いにもかかわらず、その RNase H 活性により cDNA の収量が減少します。もちろん、試薬メーカーは、cDNA の収量を増加させるために、逆転写酵素の RNase H 活性をできる限り除去するために製品を常に最適化しています。
アニーリング温度
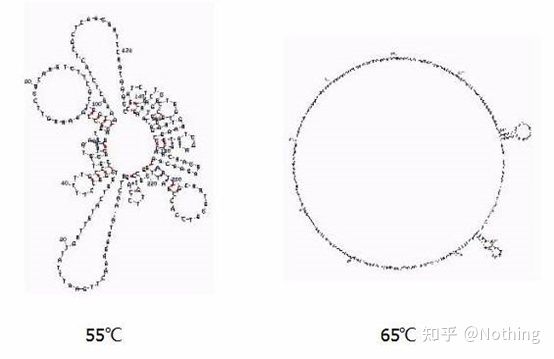
異なる温度におけるRNAの二次構造
さまざまな温度での RNA の二次構造については上の図を参照し、mFold オンライン ツールを使用して、特定の温度および塩濃度条件下でのターゲット フラグメントの二次構造を決定します。55℃では、RNAの二次構造は依然として非常に複雑で、逆転写酵素は機能せず、二次構造は65℃まで完全に解明されませんが、AMVおよびM-MLVの至適温度はこの温度よりもはるかに低くなります。
何をすべきか?二次構造は鋳型自体の相補的なペアリングであり、これによりプライマーおよび逆転写酵素と鋳型との間で強い競合が生じ、Eが低く、再現性が低いなどの一連の問題が生じます。
何をすべきか?可能な限りアニーリング温度のみを上げてください。
多くの試薬メーカーは、遺伝子工学によって逆転写酵素を改良しています。Jifan や Aidelai のように反応温度を上昇させるものや、RNase H 酵素の活性基を除去して酵素と RNA テンプレート間の親和性を向上させるものもあります。高い親和性により、二次構造を競合的に絞り出し、スムーズに読み取りを行うことができ、逆転写の効率も大幅に向上します。
ポイント:逆転写はサンプル量よりも逆転写効率(酵素が効率的であるだけでなく安定であること)の一貫性を追求することが重要であり、よほど大規模な蛍光定量PCRでないと全く実現できません。複数の cDNA。
さまざまなメーカーも、一貫性を追求するためにいくつかの努力を行っています。たとえば、現在、ほとんどの企業が逆転写反応を標準キットとしてパッケージ化して販売していますが、これは良い選択です。
たとえば、Foregene の RT Easy シリーズ キットは次のとおりです。
RT Easy I(ファーストストランドcDNA合成キット用マスタープレミックス)
MIQE (5) – 標的遺伝子情報

上の図で説明すると、
1. この遺伝子が反復実験に有効であるかどうかは、通常、反復実験によって検証できます。
2. 遺伝子 ID ですね。
3. 遺伝子長、対象遺伝子の全長は全く問題ありません。プライマーを設計するときは、増幅効率を高めるためにアンプリコンの長さが 80 ~ 200 bp であることを確認してください。
4. シーケンス Blast 比較情報。非特異的増幅を防ぐために、ターゲット遺伝子をジーンバンクで比較する必要があります。
5. 偽遺伝子の存在。偽遺伝子は、正常な遺伝子に似ていますが、正常な機能を失った DNA 配列です。それは多くの場合、真核生物の複数遺伝子ファミリーに存在します。通常はψで表されます。これは、コード遺伝子配列に非常によく似た、ゲノム内の機能しないゲノム DNA コピーです。、一般に転写されず、明確な生理学的意味はありません。
6. エクソンおよびイントロンに対するプライマーの位置。DNA コンタミネーションの問題を解決した初期の頃、私たちはプライマー、エクソン、イントロンの位置によく注意を払い、DNA の増幅を避けるためにイントロンをまたいでプライマーを設計することを一般的に検討していました。以下の図を参照してください。黒はイントロンを表し、さまざまな青はエクソンを表し、ピンクは一般的なプライマーを表し、明るい赤はイントロンにまたがるプライマーを表します。
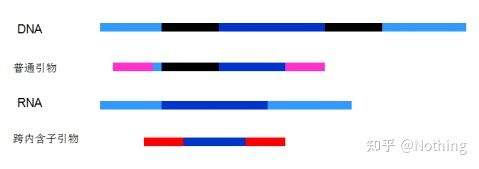
模式的、決して真実ではない
これはなんと完璧な計画のように思えますが、実際には、ほとんどの場合、トランスイントロンプライマーは想像されているほど魔法のようなものではなく、非特異的な増幅も引き起こします。したがって、DNA 汚染を防ぐ最善の方法は、DNA を完全に除去することです。
7. 立体構造の予測。この例を再度使用すると、mFold オンライン ツールを使用して、特定の温度と塩濃度でのターゲット フラグメントの二次構造を決定します。
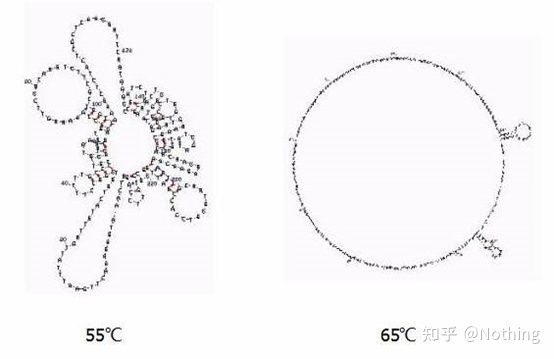
異なる温度におけるRNAの二次構造
二次構造は鋳型自体の相補的なペアリングであるため、プライマーと鋳型のペアリングの間で強い競合が発生し、プライマーが結合する機会が少なくなり、Eが低い、再現性が低いなどの一連の問題が発生します。ソフトウェア予測により、二次構造の問題がなければ、それは素晴らしいことです。存在する場合は、次の記事でこの問題の解決方法について具体的に説明します。
MIQE (6) - qPCR オリゴヌクレオチド

蛍光定量 PCR の場合、毎日苦労するのは RNA 抽出であり、次に苦労するのはプライマー設計かもしれません。
まず第一に、私たちは MIQE チェックリストに従ってプライマー設計に関するルールを確認します。クソ野郎が笑ってしまうほど簡単で、プライマープローブの配列と位置、修飾方法を調べるだけの一言で終わります。プライマー精製方法については、現在、プライマー合成は非常に安価であり、qPCR は PAGE 以上の精製方法に値し、合成機器の情報は重要ではありません。多くの人は何十年も入門書を作成していますが、そのシンセサイザーが ABI3900 であることを知りません。
プライマー設計の原則については、これらの問題はほとんどのプライマー設計ソフトウェアまたはオンライン ツールで対処できるため (推奨オンライン ツール primer3.ut.ee/)、丸暗記する必要はありません。また、プライマー設計の 99.999% は手動で行われるわけではありません。著者は 1 日に何百ものプライマーを設計することもあります。1 つずつ読むと寄り目になってしまいます。
プライマーを設計した後は、次の点を確認してください。
1. プライマーを 3' 末端近くに設計する:cDNA ファーストストランド合成にオリゴ dT プライマーを使用する場合、逆転写効率と RNA の完全性を考慮して、増幅効率を向上させるために、設計するプライマーを 3' 末端近くに設計する必要があります。絵を使って次のように説明します(これを理解する方法はありません)。
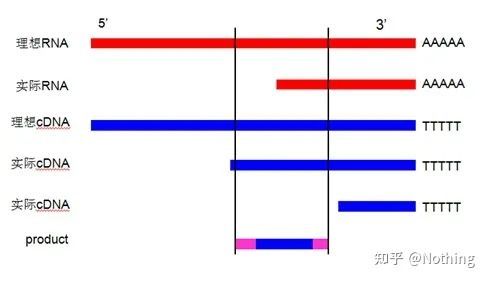
なぜプライマーを 3' 末端近くに設計する必要があるのか、それは真実ではないはずです
2. TM 値: Tm 値は 55 ~ 65 ℃ (エキソヌクレアーゼ活性は 60 ℃ で最も高くなるため)、GC 含量は 40% ~ 60% です。
3. BLAST: ゲノムの非特異的増幅を回避するために、補足検証には Blast を使用する必要があります。
MIQE(7) - qPCR プロセス
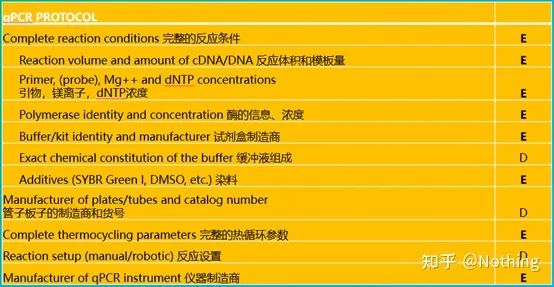
1. qPCRキット
MIQEの要件に従い、PCR反応系の構成、使用したキット、メーカー、反応系の大きさ、色素法かプローブ法のどちらを使用するか、PCRプログラムの設定など、完全な反応条件を論文に明確に記載する必要があります。ベテランドライバーならキットさえ選べば基本的に上記の情報で決まることが分かると思います。
現在、蛍光定量 PCR キットの製造と生産は非常に成熟した技術です。よほど悪質なメーカーを選択しない限り、問題が発生する可能性は高くありませんが、それでもいくつかのポイントを共有したいと思います。
ホットスタート Taq 酵素:PCR の最も重要な部分はホットスタート Taq 酵素です。市販されているホットスタート酵素は大きく2種類に分けられ、1つは化学的に修飾されたホットスタート酵素(パラフィン包埋と想像していただけます)、もう1つは抗体修飾(抗原抗体結合)用のホットスタート酵素です。化学修飾は酵素をホットスタートさせる初期の方法です。一定の温度に達すると酵素は活性を発揮します。抗体修飾ホットスタート酵素は、生物学的手法を使用して酵素の活性をブロックします。抗体は一定の温度に達すると変性してタンパク質として失活し、酵素活性が働きます。
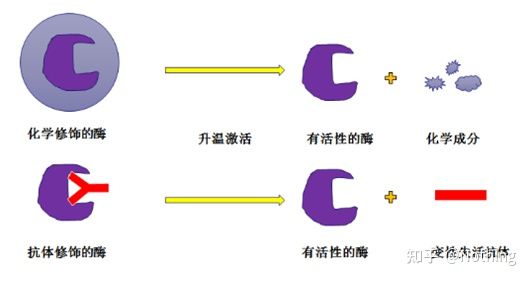
しかし、これは何の役に立つのでしょうか?これは、抗体修飾酵素の方が化学修飾酵素よりも放出活性が速いため、感度の点では抗体修飾酵素の方が若干有利であり、市販のキットには基本的に化学修飾酵素は入っていません。もしそうなら、このメーカーの技術はまだミレニアムの時代に留まっているということになります。
マグネシウムイオン濃度:マグネシウムイオン濃度は PCR 反応において非常に重要です。適切なマグネシウムイオン濃度は、Taq 酵素活性の放出を促進します。濃度が低すぎると、酵素活性が大幅に低下します。濃度が高すぎると、酵素触媒による非特異的増幅が増強されます。マグネシウムイオンの濃度は、プライマーのアニーリング、テンプレートおよび PCR 産物の融解温度にも影響し、それによって増幅フラグメントの収量に影響します。マグネシウムイオンの濃度は通常25mMに制御されます。もちろん、良いキットを作るには、マグネシウムイオンの濃度を適切に制御する必要があります。一部の販売業者は、試薬にマグネシウムイオンキレート剤を添加しており、これによりマグネシウムイオン濃度の自動調整の効果が得られます。
蛍光色素濃度:私たちが通常使用するSYBR Greenである蛍光色素は、主に二本鎖DNAの副溝に結合することによって蛍光を発生します。これは、二本鎖DNAへの色素の結合が非特異的であるためです。つまり、二本鎖DNAが結合している限り蛍光が発生する可能性があるため、システム内のプライマーダイマーとDNAテンプレートが結合してバックグラウンドシグナルを形成します。
PS: 光に敏感な性質があるため、市販の製品は通常、茶色の不透明な遠沈管にパッケージされています (下の写真を参照)。ただし、これでは問題が発生します。サンプリング時に液体が吸い込まれているかどうかが分かりにくい。この点では、Qingke が最も使いやすく (下の写真に示すように)、透明なチューブが不透明な缶の袋に梱包されています。次に、光を避けてサンプリングする利便性を考慮して、缶の袋に入れます。正しい製品番号を選択する必要があります。TSE204は草を植えたくなる超コストパフォーマンスの存在です。

蛍光色素の濃度も非常に重要です。濃度が低すぎると、後の段階で増幅曲線が上がらず、完全ではなくなります。濃度が高すぎるとノイズ干渉の原因となります。蛍光定量 PCR は主に CT 値に依存するため、蛍光色素の濃度が適切に調整されていない場合、高い点よりも低い点の方が優れています。もちろん、適切な染料濃度が最適です。
ロックス: ROX 色素は、ウェル間の蛍光シグナル誤差を補正するために使用されます。一部の機器メーカーは校正を必要としますが、その他の機器メーカーは校正を必要としません。たとえば、Thermo Fisher Scientific のリアルタイム PCR 増幅装置を使用するには、通常、7300、7500、7500Fast、StepOnePlus などの校正が必要です。これについては、一般的なキットの説明書に記載されています。
Foregene の qPCR Mix には、さまざまなモデルでの使用に便利な ROX 色素も含まれています。
弱水素結合処理: 弱い水素結合の処理は比較的技術的な問題です。多くのキットのマニュアルを読んだことはありませんが、そのどれもこのトピックについて言及していませんでした。実際、それはとても重要です。塩基の組み合わせは主に水素結合の強さに依存します。強い水素結合は通常の増幅であり、弱い水素結合は非特異的な増幅を引き起こします。弱い水素結合をうまく除去できない場合、非特異的な増幅が避けられません。著者の範囲内では、この問題に気づいている企業はわずかです。キットを購入する際には、選択したいキットに対してこの点の解決策を検討しているかどうかを参考にすることができます。
反応量: 20 ~ 50ul システムがより一般的に使用され、これより少ない量ではエラーが発生する可能性があります。一般的に、キットの説明書では PCR 反応量の使用を推奨しています。コストを節約するために、賢明に小さいボリュームを使用しないでください。の目標。販売店が推奨する容量は実際にテストしたものであり、少量による誤差の問題は解決できない可能性があります。
2. 管板のメーカーと品番
蛍光定量 PCR の原理は誰もが知っています。蛍光収集は主に PCR チューブ キャップを通じて行われます。PCR 消耗品を選択する際は、光透過性が良いこと、装置に適していることの 2 点に注意してください。基本的には主流ブランドの板や管で問題ありませんが、適応性を考慮して慎重に選ばないと楽器を使用できなくなります。

4. トップレベルの知識
MIQE (8) - qPCR 検証
これが qPCR の最優先事項です。非常に多くの英雄がここで砂に落ちました。もちろん、あなたが幸運で、研究した遺伝子が単純だったので、風に乗って氷の洞窟を漂っていた可能性もあります。qPCR の検証情報は、データの信頼性をテストすることを目的としています。必要な検証情報は次のとおりです。
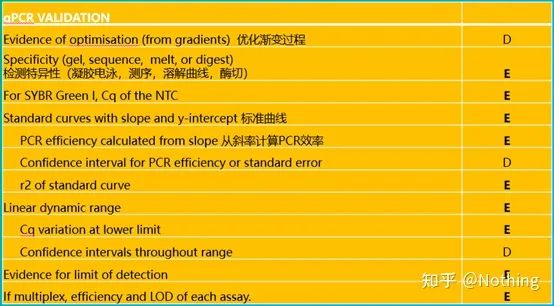
1.特異性試験
標的遺伝子増幅の特異性は、電気泳動画像が単一のバンドであるかどうかを確認することによってテストされます。配列の検証。融解曲線でピークマップが単一かどうかを確認します。酵素消化の検証やその他の方法。
ここでは、t に焦点を当てます。融解曲線法を用いた非特異的増幅の分析。一般的に、プライマーを設計する場合、産物フラグメントのサイズは 80 ~ 200 bp の範囲である必要があり、PCR 産物の融解温度は 80 ~ 85 °C の範囲になります。したがって、雑多なピークがある場合は、他の非特異的な増幅産物が存在するはずです。ピークが 80°C 未満で現れる場合、それは一般にプライマーダイマーであると考えられます。ピークが 85°C を超えて現れる場合は、一般に DNA 汚染または大きな断片の非特異的増幅であると考えられます。
注: 場合によっては、80°C にピークが 1 つだけ存在することがあります。現時点では、この概念を遵守する必要があります。おそらく、増幅結果はすべてプライマーダイマーであると考えられます。
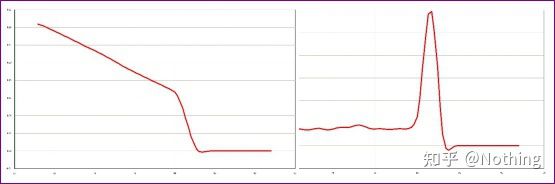
通常の融解曲線 (非特異的増幅のない単一ピーク)
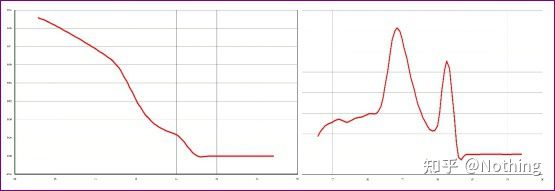
問題のある融解曲線 (スプリアスピークの非特異的増幅)
【事例分析】
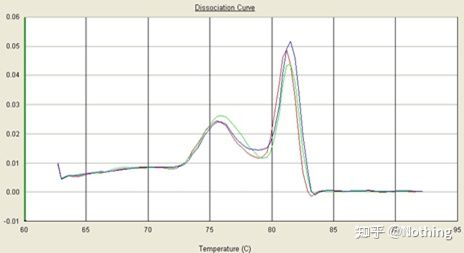
メインピークはあるがプライマーダイマーが深刻
以下の図の単一ピークの融解曲線は、完璧な実験であると簡単に目を欺きますが、結果は完全に間違っています。このとき、融解温度を確認する必要があります。ピーク温度は 80°C 以下で、完全にプライマー-ダイマーです。
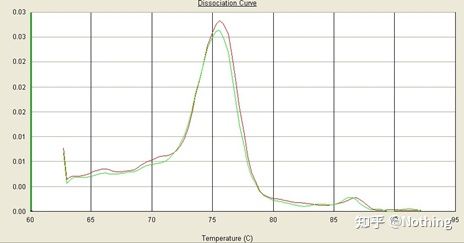
ターゲットフラグメントなし、すべてのプライマーダイマー
ここで兄は止まらない。下の写真は、クソ野郎から送られてきた携帯電話で撮った写真です。彼が使用した試薬はすべて業界で一般的に使用されているブランドです。彼はある T プレフィックス ブランドから別の T プレフィックス ブランドに変更しました。もうお察しだと思います。クソ野郎は私にこう叫びました。「最初の写真で使用した試薬は良すぎて、ピークは単一です。その後、お勧めしていただいた試薬を使用すると、2枚目の写真のようにピークが混在した状態になります。あなたは私を惨めにしました。「
2 つのグラフを分離します。一見すると、一方は単一のピークを持ち、もう一方は二重ピークを持ちます。ナンセンス、もちろん単一のピークでも問題ありません。本当?
Dou Eよりもひどいのは、下の2枚の写真を並べてみればすぐに分かるだろう。実際、私たちはこの種のイメージに簡単に麻痺してしまいます。注意深く分析した結果、次のことがわかりました。最初の図のピークは 75℃ であり、これは完全にプライマー二量体です。2番目の図のピークは75℃と82℃に現れ、少なくとも生成物が現れます。
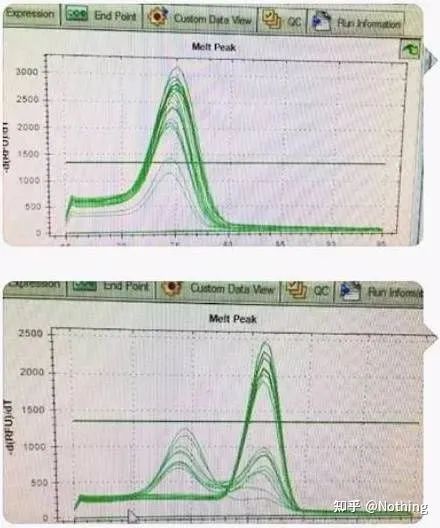
生徒からのフィードバックの写真
したがって、根本的な問題は試薬の問題ではなく、プライマーの設計の問題です。同時に、これは一部の大手ブランドが鉄の品質に問題があることも証明しており、私の兄が前に言っていたこと、つまりあなたの論文を支持しているのは試薬ブランドではないことも証明しています。試薬のブランドを押し上げたのはあなたの記事です。想像してみてください。もしこの最低な奴が試薬を変更しなかったら、間違ったデータがジャーナルに送信され、悲劇が起こるでしょう。
2. ブランクコントロールのCt値
説明しないでください、ブランクコントロールにCt値がある場合、それは汚染ではありませんか?ただし、どのブランク コントロールに Ct 値があるかを理解する必要があります。NTCであれば試薬の混入などの外来DNAがあることを意味します。NRTの場合は、抽出したRNAにDNAが混入していることを意味します。
3. 標準曲線
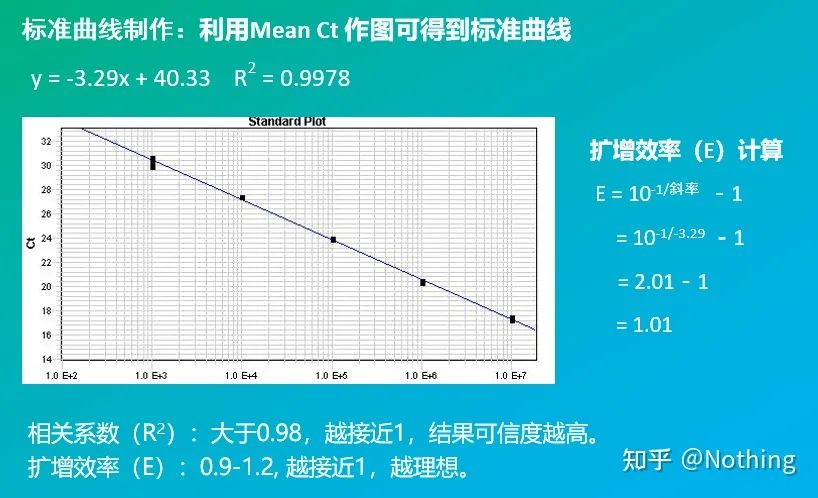
傾きと計算式を含めて、PCR 効率は計算式によって計算できます。完璧な実験を行うには、標準曲線の傾きが 3.32 に近づき、R² が 0.9999 に近づく必要があります。
4. リニアダイナミックレンジ
反応のダイナミックレンジは線形です。標準曲線の作成に使用したテンプレートに応じて、ダイナミック レンジには少なくとも 5 つの濃度勾配が含まれ、高濃度勾配と低濃度勾配での Ct 値の変化に注意する必要があります。
5. 検出精度
qPCR 結果の変化、つまり再現性の低下、つまり精度の低下は、温度、濃度、操作などの多くの要因によって引き起こされます。qPCR の精度は一般に、コピー数が減少するにつれて制御しにくくなります。理想的には実験内変動であり、この技術的変動は生物学的変動とは区別されるべきであり、生物学的複製はグループ間または治療間の qPCR 結果の統計的差異に直接対処できます。特に診断アッセイの場合、施設およびオペレーター全体で最高のアッセイ間精度 (再現性) を報告する必要があります。
6. 検出効率と LOD (マルチプレックス qPCR における)
LOD は、検出された陽性サンプルの 95% の最低濃度です。言い換えれば、標的遺伝子複製セット内に含まれる LOD の濃度は、失敗した反応の 5% を超えてはなりません。マルチプレックス qPCR 分析を行う場合、特に点突然変異や多型を同時に検出する場合、マルチプレックス qPCR は、同じチューブ内で複数のターゲット フラグメントの精度が損なわれていないという証拠を提供する必要があり、複数の検出と単一チューブの検出の効率と LOD が同じである必要があります。特に、高濃度の標的遺伝子と低濃度の標的遺伝子を同時に増幅する場合には、この問題に注意が必要である。
問題と解決策一般に、qPCR デバッグでよく発生する問題は、次の側面に焦点を当てています。
・非特異的増幅
・プライマー濃度選択の難しさとプライマーダイマーの問題
・アニール温度が不正確である
・増幅効率に影響を与える二次構造
非特異的増幅
非特異的増幅が発生する場合は、プライマーの設計が適切でないことが一般的に考えられますが、プライマーを変更することを急いでいない場合は、まず次の方法を試すことができます(原則も添付されています)。
·アニーリング温度を上げる – 弱い水素結合を維持できなくなるようにします。
·アニーリングと伸長時間を短縮 – 弱い水素結合の可能性を減らします。
·プライマー濃度を下げる – 重複したプライマーと非ターゲット領域が結合する可能性を減らします。
増幅効率が低い
非特異的増幅とは逆の状況 – 低い増幅効率、および低い増幅効率に対処するための対策は、まさにその逆です。
·焼きなましと伸びの時間を延長します。
· 3 ステップ PCR に変更し、アニーリング温度を下げます。
・プライマー濃度を上げる。
Ps: 90年代生まれの大学院生の多くは、実験のデバッグ方法を勉強する気はなく、キットが問題を完全に解決できることを望んでいます(卒業後、試薬会社に行って研究開発をしたい場合)。実際、試薬メーカーもこのように考えています。愚か者であってほしいと願っていますが、入手できればすぐに使用できるため、試薬メーカーは弱い水素結合吸収因子の導入を含め、非特異的増幅の問題を解決するために多大な努力を費やしてきました。この問題を簡単に解決するには、愚か者は依然として試薬会社の紹介文を読んで、弱い水素結合を吸収する因子があるかどうかを確認する必要があります。
プライマー濃度の選択が難しく、プライマーダイマーに関する問題が発生する
方法 1: 一般に、qPCR 用のキットの説明書には、推奨システムと推奨プライマー濃度が記載されています。
方法 2: プライマーの濃度勾配を設定してデバッグします。下の写真は説明のためにある会社から盗んだものです。下の図は、3 つのプライマー濃度勾配 (100nM、250nM、500nM) と 4 つのテンプレート濃度勾配 (0.1ng、1ng、10ng、100ng) で作成された蛍光定量結果を示しています。実験結果の Ct 値は次のようにプロットされます。
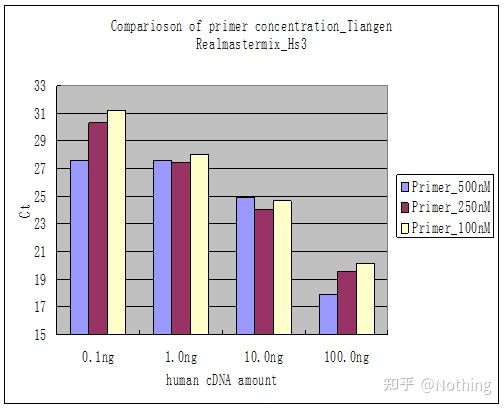
プライマー濃度の選択 次のように、各プライマー濃度を 1 行に連結します。
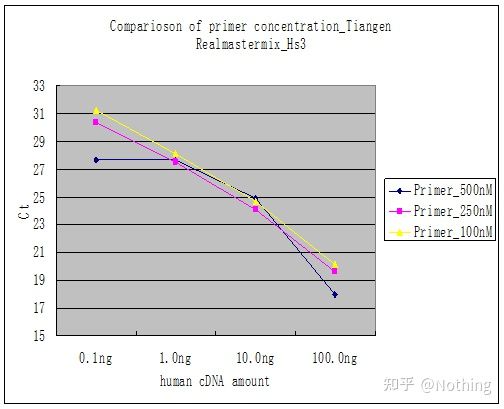
プライマー濃度の選択は明らかで、100nM と 250nM のプライマー濃度の直線関係はより良好であり、500nM のプライマー濃度の直線関係は比較的悪いです。100nM および 250nM では、250nM の Ct 値は比較的小さいため、最適なプライマー濃度は 250nM です。一般に、深刻なプライマーダイマーが融解曲線に見られます。設計されたプライマーがプライマーダイマーを回避できない場合はどうなるでしょうか?
方法 3:プライマーの量を減らし、アニーリング温度を上げます(説明する必要はありません)。
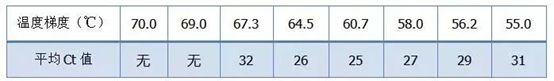
アニーリング温度の経験値は 60°C です。よくわからない場合、より適切なアニーリング温度を選択するにはどうすればよいですか?答えはプライマー濃度の選択と同じです。勾配テスト。問題を説明するために、Bio-rad 社から写真を入手してください。特定のターゲットフラグメントを増幅する場合、8 つの温度勾配を設定し、それぞれを 3 回繰り返します。得られる増幅曲線は次のとおりです。
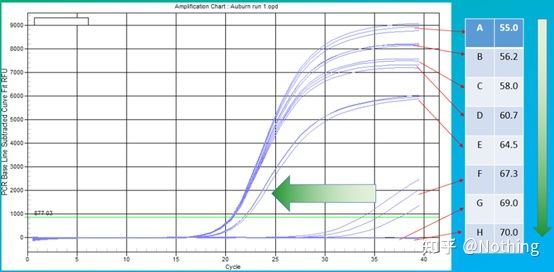
アニーリング温度の選択:
・70°C、69°C - 基本的にプライマーは結合できないため、増幅は行われません。
・67.3℃ – 初期の増幅が少なく、Ct値が比較的大きい。
・64.5℃——Ct値が低下します。
・60.7℃、58.0℃、56.2℃、55.0℃では、基本的にCt値は安定する傾向にありましたが、最終的な蛍光値は異なりました。
選び方は?原則: 第一の原則は、Ct 値が高いことです。同じ Ct 値の場合、二量体化と非特異的増幅を避けるために、より高いアニーリング温度を選択します。55℃では蛍光値が高くなりますが、二量体または非特異的増幅が存在する可能性があります。
しかし、あなたと同じくらい賢い人なら、次のように考えるはずです。論理的に言えば、PCR 反応が非常に特異的である場合、プライマー濃度が最小要件を超えている限り、蛍光色素や dNTP と同様に、高点と低点は影響を及ぼさないはずです。実際、アニーリング温度が適切に最適化されている限り、Ct 値に対するプライマー濃度の影響は当然最小限に抑えられます。
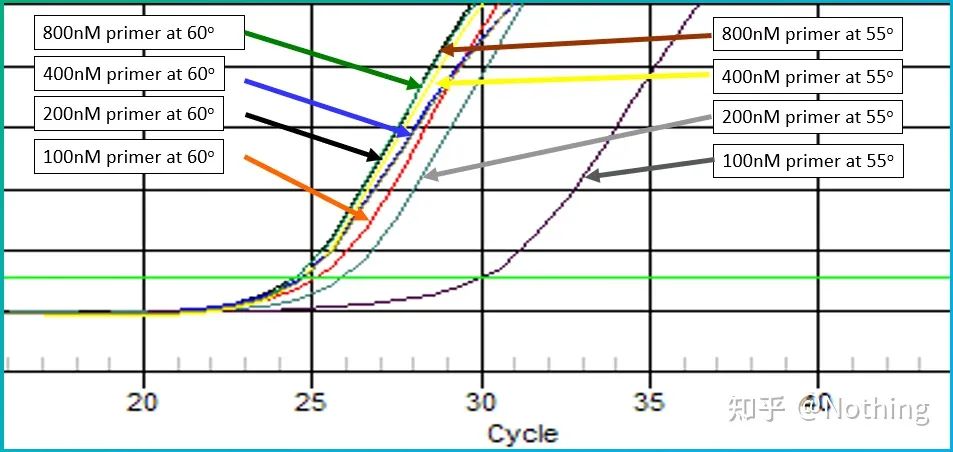
アニーリング温度が適切に最適化され、プライマー濃度によるCTへの影響が最小限に抑えられます。
二次構造が増幅効率に影響する
問題を説明するために Bio-rad から写真を取り出してみましょう。また、二次構造を持つ遺伝子を増幅するための温度勾配も設計します。
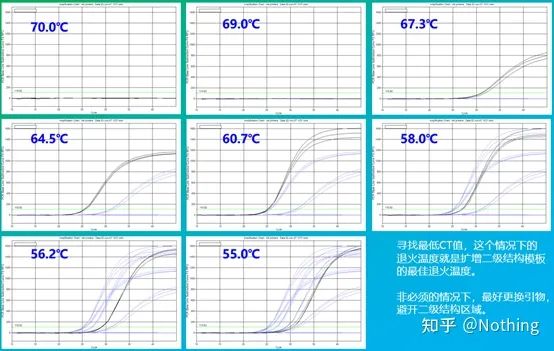
二次構造が現れる
温度勾配が減少するにつれて生成物が出現し始め、Ct値が上昇し、60.7℃で最小値に達し、その後温度勾配が減少するにつれてCt値が大きくなっていることがわかります。逆に、温度が上昇すると二次構造が開き、増幅効率が増加します。一定の温度に達した後、温度を上げても増幅効率は向上しません。この時点ではプライマーを安定して組み合わせることができないためです。したがって、Ct値が最も低い温度を探す、これは二次構造テンプレートを増幅するのに最適な温度です。もちろん、賢明な愚か者は、必要がない場合はプライマーを変更し、二次構造領域を避けるのが最善であることを知っているはずです。
5. アプリケーションレベル
MIQE—データ分析

データ分析は主に蛍光定量 PCR 装置によって行われます。前回の記事では、実験計画で説明したブランク コントロールなど、多くのデータ分析作業を行いました。内部参照遺伝子、リピート数等が明らかになりました。, ここでは主にqPCRの応用について説明します。
qPCR は広く使用されており、実験的検証と核酸診断が最も一般的に使用されるシナリオです。
絶対定量化
Log (初期濃度) はサイクル数と線形関係があります。標準曲線は、既知の初期コピー数をもつ標準から描くことができます。つまり、増幅反応の直線関係を得ることができます。試料のCt値に応じて、試料中の濃度を計算することができます。含めるテンプレートの数。
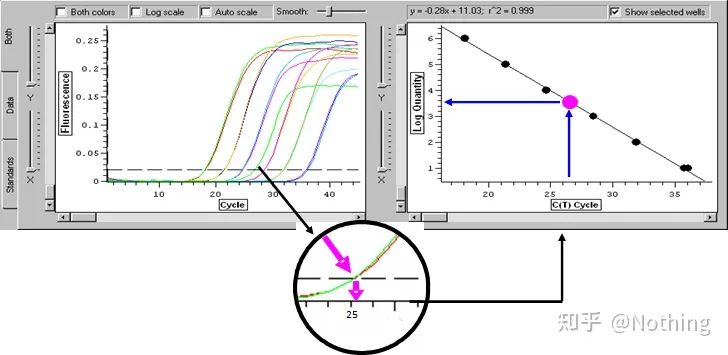
絶対定量計算法
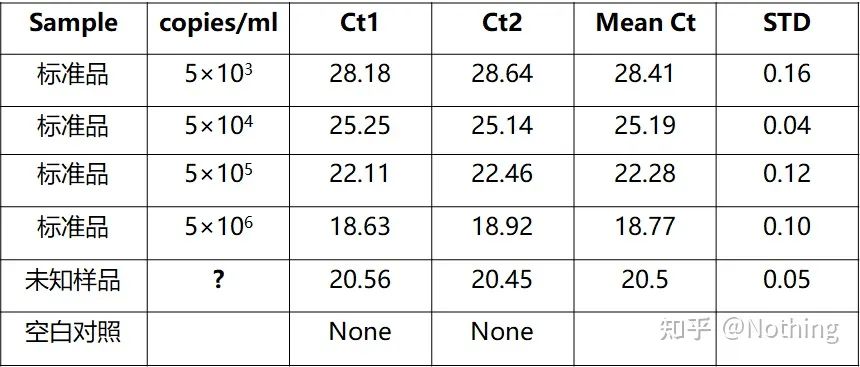
絶対定量は標準曲線に基づく必要があります。検量線を作成するには、標準が必要です。通常、スタンダードは目的遺伝子をクローニングして得られるプラスミドです。なぜプラスミドなのか?環状プラスミド DNA が最も安定しているためです。標準品を2倍(10倍希釈)に応じて5~6段階に希釈し、均一性に注意して希釈してください。Ct 値を 15 ~ 30 の範囲内にします。
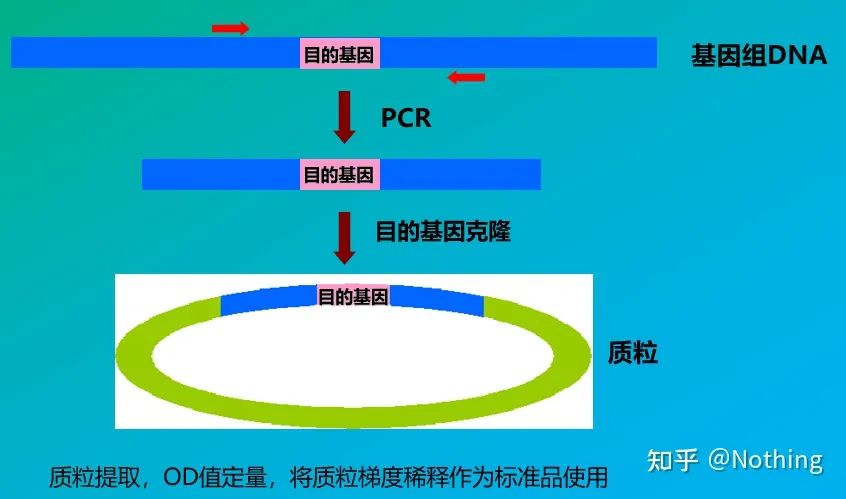
標準的な調製
同時に、テストするサンプルもそれに応じて希釈する必要があり (希釈係数を忘れないでください)、Ct 値も 15 ~ 30 の間に収まる必要があります。標準品と被検査サンプルを一緒に装置にセットします。分析後、標準物質を用いて検量線を作成し、その検量線に被験サンプルを当てはめて濃度を算出した。
B型肝炎ウイルスHBV定量は代表的な絶対定量であり、血液1ml中のウイルスコピー数を計算できます。
コピー数の計算
試験するサンプル濃度 (ng/ul) = OD260 × 50ug/ml × 希釈率
サンプルの分子量=塩基数×324
被検サンプルのコピー数(コピー/μl)=被検サンプルの濃度/サンプルの分子量×6×1014

コピー数の計算方法

以上が数量を求める計算方法となります。これは中学校を卒業すれば解ける数学の問題であり、数学の問題は一般にコンピュータによって解けます。理解できない場合は、コミュニケーションをとることができます。
相対定量化
相対定量化は主に科学研究で使用されます。1 ml の血液中に何個のウイルスが存在するか、そしてそれは DNA ウイルスですが、これは比較的決定的な事象です。血液の量は決定可能であり、DNA ウイルスは比較的安定しています。しかし、葉の大きさ、重さ、柔らかさを測定することが難しく、抽出されたRNAの量を測定することが難しく、逆転写の効率を測定することも難しいため、葉内の特定の遺伝子の転写コピー数を比較することは困難であり、つまり、どのステップでも実験データにバグが発生し、使用できない可能性があります。
したがって、相対定量化では次の要素を導入する必要があります。内部参照遺伝子 。
言い換えれば、相対定量化は実際には、標的遺伝子と内部参照遺伝子との間の比較である。同一組織、同一細胞内で比較した場合、サンプルサイズ、RNA抽出量、逆転写効率、PCR効率の影響は比較的小さいです。サンプルサイズが小さいため、内部参照遺伝子と標的遺伝子の両方が比較的減少しました。これまで私たちが均一性と安定性を重視してきたのはこのためです。
内部参照遺伝子は一般に、ハウスキーピング遺伝子(ハウスキーピング遺伝子)。これは、すべての細胞で安定して発現される遺伝子のクラスを指し、その産物は細胞の基本的な生命活動を維持するために必要です。
この概念を混同しないでください。ハウスキーピング遺伝子は生物学的機能用語であり、内部参照遺伝子は実験の専門用語です。ハウスキーピング遺伝子は、内部参照遺伝子として選択される前に検証に合格する必要があります。
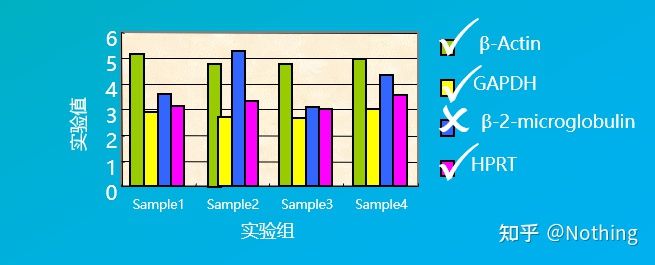
たとえば、以下の図にあるいくつかのハウスキーピング遺伝子を選択して、さまざまな組織細胞での発現レベルをテストしたところ、β-2-ミクログロブリンの発現レベルが他の 3 つの遺伝子の発現レベルと大きく異なるため、内部参照遺伝子として使用できないことがわかりました。
内部参照遺伝子の補正機能を理解した後、内部参照遺伝子の導入により 2 つのアルゴリズムが導出されます。
・二重検量線法
・2 – △△Ct法(CT値比較法)
種や遺伝子の機能を研究することに興味がある場合は、アルゴリズムの研究をあきらめて数式を直接使用するか、機械を直接使用してください。もしあなたが数学と工学に精通している人なら、お気軽にどうぞ。
二重標準曲線法
対照サンプルおよび被検サンプルの標的遺伝子およびハウスキーピング遺伝子を検量線により定量し、計算式に従って相対値を算出したものが相対発現量となります。
利点: 分析が簡単、実験による最適化が比較的簡単
欠点: 遺伝子ごとに、実験の各ラウンドで標準曲線を作成する必要があります。
用途: 遺伝子発現制御の研究において最も一般的に使用され、認知されている 2 つの相対定量的手法のうちの 1 つ
式は次のとおりです。

例は次のとおりです。
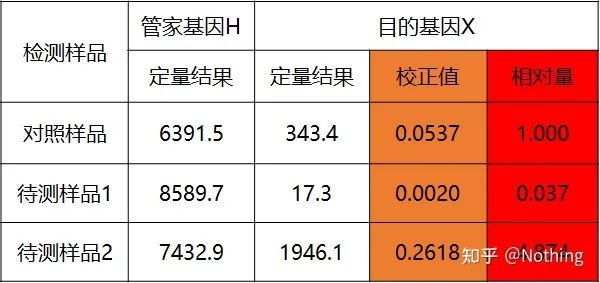
定量結果に基づいて相対量を計算
2 – △△Ct法(CT値比較法)

利点: 標準曲線を作成する必要がない
短所: 増幅効率は 100% に近いと想定されます。標準偏差は < 5% であり、標準曲線と各増幅間の効率は一致していると想定されます。実験条件の最適化はさらに複雑です。
用途: 遺伝子発現制御の研究において最も一般的に使用され、認知されている 2 つの相対定量的手法のうちの 1 つ
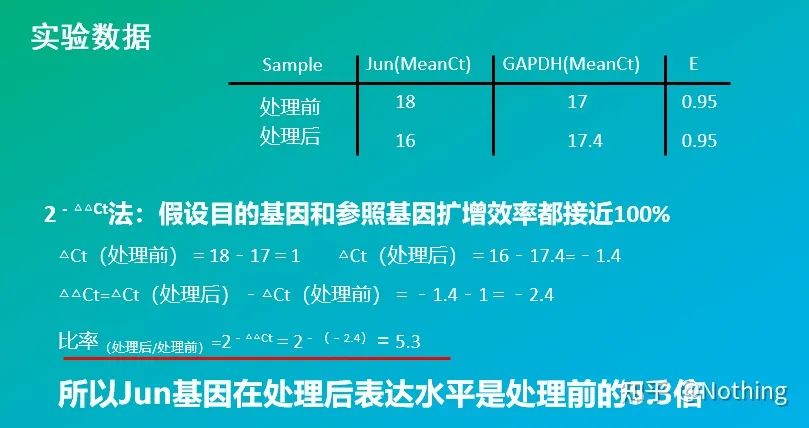
もちろん、増幅効率を完璧にすることは通常不可能です。 1. 補正方法:ターゲット遺伝子とリファレンス遺伝子の増幅効率が同じであることがわかっていても、増幅効率が 1 に等しくない場合、2-△△Ct は (1+E )-△△Ct のように補正できます。たとえば、増幅効率が 0.95 の場合、計算式は 1.95-△△Ct と補正できます。
ここまでで蛍光定量PCRに関する内容は終了です。
投稿時間: 2023 年 4 月 6 日



