



RT-qPCR は通常の PCR 技術を発展させたものです。従来の PCR 反応システムに蛍光化学物質 (蛍光色素または蛍光プローブ) を追加し、異なる発光メカニズムに従って PCR のアニーリングおよび伸長プロセスをリアルタイムで検出します。培地中の蛍光シグナルの変化は、PCR の各サイクルでの産物の変化量を計算するために使用されます。現在、最も一般的な方法は蛍光色素法とプローブ法です。
蛍光色素法:
SYBR Green Ⅰ、PicoGreen、BEBO などの一部の蛍光色素は、それ自体では発光せず、dsDNA の副溝に結合して蛍光を発します。したがって、PCR 反応の開始時には、機械は蛍光シグナルを検出できません。反応がアニーリング-伸長段階(2段階法)または伸長段階(3段階法)に進むと、この時点で二本鎖が開き、新たなDNAポリメラーゼ鎖合成中にdsDNA副溝に蛍光分子が結合して蛍光を発します。PCR サイクル数が増加するにつれて、より多くの色素が dsDNA と結合し、蛍光シグナルも継続的に強化されます。SYBR Green Ⅰを例に挙げます。
プローブ方法:
Taqman プローブは、最も一般的に使用される加水分解プローブです。プローブの 5' 末端には蛍光基があり、通常は FAM です。プローブ自体は、標的遺伝子に相補的な配列である。蛍光色素分子の 3' 末端には蛍光消光基があります。蛍光共鳴エネルギー移動(フェルスター共鳴エネルギー移動、FRET)の原理によれば、レポーター蛍光基(ドナー蛍光分子)と消光蛍光基(アクセプター蛍光分子)の励起スペクトルが重なり、その距離が非常に近い(7~10nm)場合、ドナー分子の励起によってアクセプター分子の蛍光が誘導される一方、自己蛍光は弱められます。したがって、PCR 反応の開始時に、プローブがシステム内で遊離しており無傷である場合、レポーター蛍光基は蛍光を発しません。アニーリングの際、プライマーとプローブはテンプレートに結合します。伸長段階では、ポリメラーゼは継続的に新しい鎖を合成します。DNA ポリメラーゼは 5'-3' エキソヌクレアーゼ活性を持っています。プローブに到達すると、DNA ポリメラーゼはテンプレートからプローブを加水分解し、レポーター蛍光基をクエンチャー蛍光基から分離し、蛍光シグナルを放出します。プローブとテンプレートは 1 対 1 の関係にあるため、検査の精度と感度の点でプローブ法の方が色素法よりも優れています。
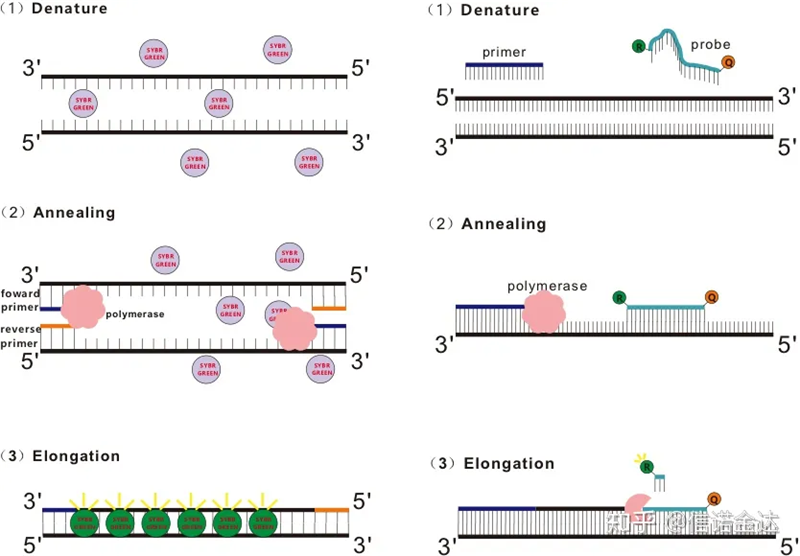
図1 qRT-PCRの原理
プライマー設計
原則:
プライマーは、核酸シリーズの保存領域内に設計され、特異性を持つ必要があります。
cDNA配列を使用するのが最適ですが、mRNA配列も使用できます。そうでない場合は、DNA 配列の cds 領域の設計を調べます。
蛍光定量産物の長さは 80 ~ 150 bp、最長は 300 bp、プライマーの長さは通常 17 ~ 25 塩基であり、上流プライマーと下流プライマーの差が大きすぎてはなりません。
G+C 含有量は 40% ~ 60% で、45 ~ 55% が最適です。
TM 値は 58 ~ 62 度です。
プライマーダイマーとセルフダイマーを避けるようにしてください (連続した相補的塩基が 4 対を超えて現れないようにします) ヘアピン構造、やむを得ない場合は、ΔG<4.5kJ/mol* にする 逆転写クリーン中に gDNA が除去されたことを確認できない場合は、イントロンのプライマーを設計するのが最善です *3' 末端は修飾できません。また、AT、GC リッチ領域を避けるために、T/C、A/G 連続構造 (2-3) プライマーおよび非
特異的 不均一に増幅された配列の相同性は、好ましくは70%未満であるか、または8つの相補的塩基相同性を有する。
データベース:
CottonFGD キーワードで探す
プライマー設計:
IDT-qPCRプライマー設計
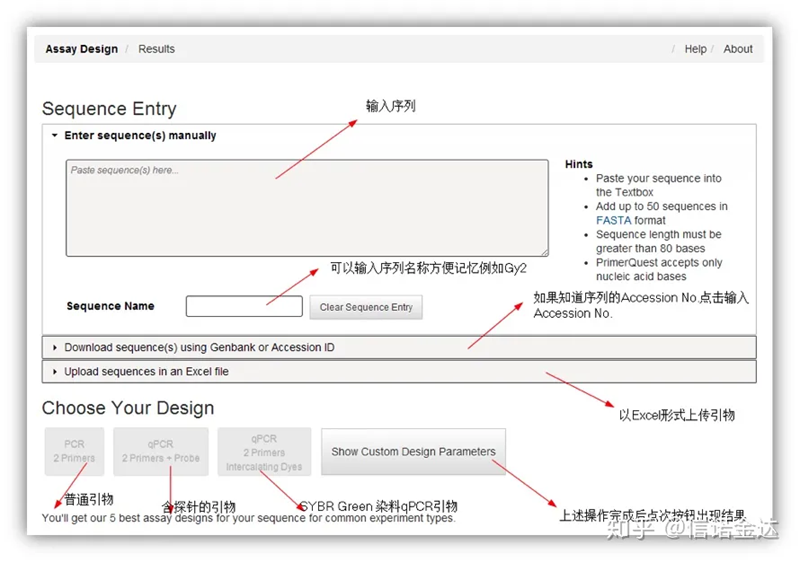
図2 IDTオンラインプライマー設計ツールページ
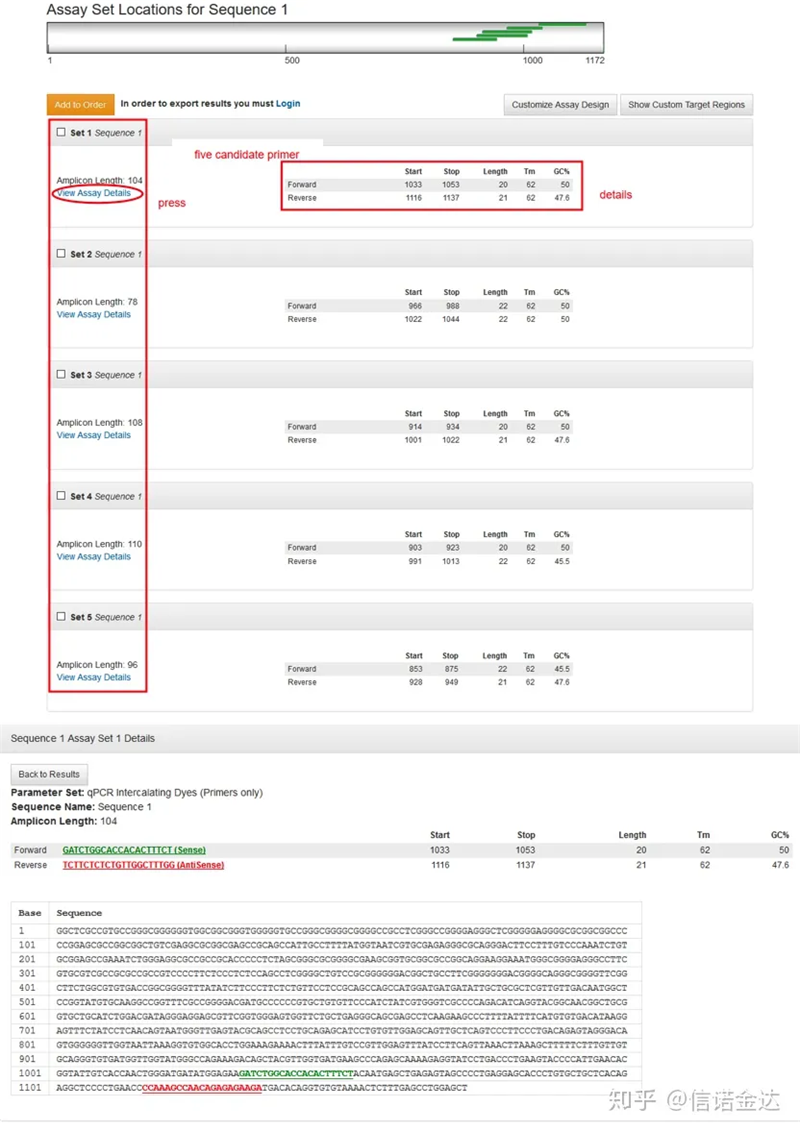
図3 結果ページの表示
lncRNAプライマーの設計:
lncRNA:mRNAと同じ手順です。
miRNA:ステムループ法の原理: すべての miRNA は約 23 nt の短い配列であるため、直接 PCR 検出を行うことができないため、ステムループ シーケンス ツールが使用されます。ステムループ配列は約50ntの一本鎖DNAであり、単独でヘアピン構造を形成することができます。3 '末端を miRNA の部分断片に相補的な配列として設計することで、逆転写時に標的 miRNA をステムループ配列に接続することができ、その全長は 70 bp に達することができ、これは qPCR によって決定された増幅産物の長さと一致します。テーリング miRNA プライマーの設計。
増幅特異的検出:
オンライン爆発データベース: 配列類似性による CottonFGD ブラスト
ローカル ブラスト: ローカル ブラストを行うには Blast+ の使用を参照してください。Linux と MacOS はローカル データベースを直接確立できます。Win10 システムは ubuntu bash をインストールした後に行うこともできます。ローカル blast データベースとローカル blast を作成します。win10 で ubuntu bash を開きます。
注: 陸地綿と海島綿は 4 倍体作物であるため、発芽の結果は 2 つ以上一致することがよくあります。これまでは、NAU cd をデータベースとして使用して blast を実行すると、SNP がわずかに異なるだけで 2 つの相同遺伝子が見つかる可能性がありました。通常、2 つの相同遺伝子はプライマー設計では分離できないため、同じものとして扱われます。明らかなインデルがある場合、通常、プライマーはインデルに基づいて設計されますが、これによりプライマーの二次構造が発生する可能性があり、自由エネルギーが高くなり、増幅効率が低下しますが、これは避けられません。
プライマー二次構造の検出:
手順:オリゴ 7 を開く → テンプレート配列を入力 → サブウィンドウを閉じる → 保存 → テンプレート上でプライマーを見つけ、ctrl+D を押してプライマーの長さを設定 → 自己二量体、ヘテロ二量体、ヘアピン、ミスマッチなどのさまざまな二次構造を解析します。 図 4 の最後の 2 つの写真は、プライマーのテスト結果です。フロントプライマーの結果は良好で、明らかなダイマーおよびヘアピン構造はなく、連続した相補的塩基はなく、自由エネルギーの絶対値は 4.5 未満でしたが、バックプライマーは連続した 6 塩基が相補的で、自由エネルギーは 8.8 でした。さらに、より深刻な二量体が 3' 末端に現れ、4 つの連続した塩基の二量体が現れます。自由エネルギーは高くありませんが、3' ダイマー Chl は増幅特異性と増幅効率に重大な影響を与える可能性があります。さらに、ヘアピン、ヘテロダイマー、およびミスマッチを確認する必要があります。
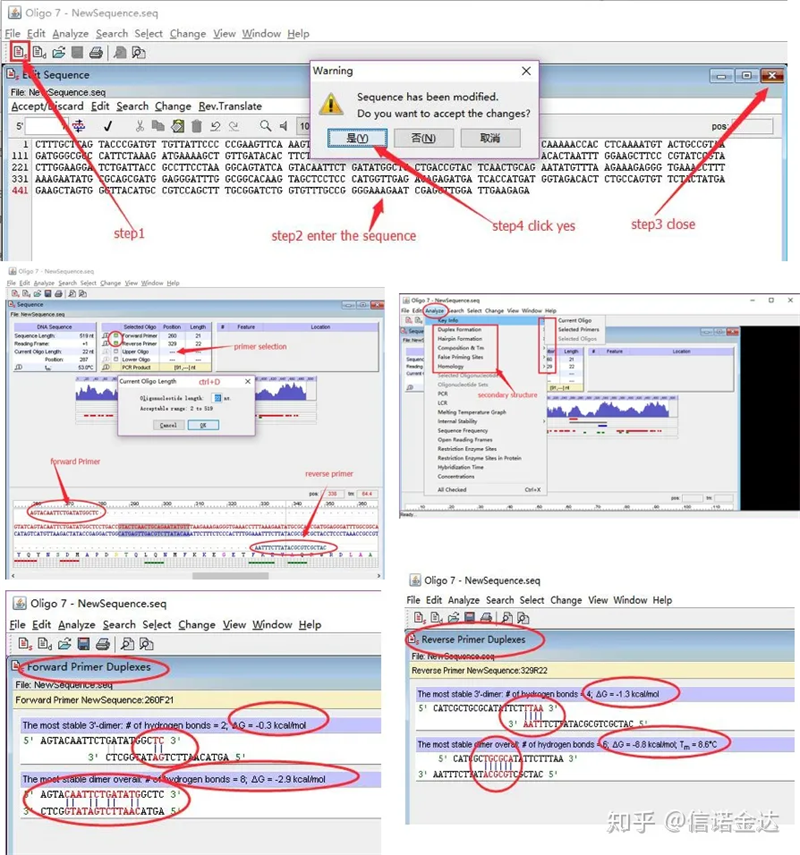
図3 oligo7の検出結果
増幅効率の検出:
PCR 反応の増幅効率は PCR の結果に重大な影響を与えます。qRT-PCR においても、増幅効率は定量的な結果にとって特に重要です。反応バッファー内の他の物質、機械、プロトコルを除去します。プライマーの品質も qRT-PCR の増幅効率に大きく影響します。結果の精度を確保するには、相対蛍光定量と絶対蛍光定量の両方でプライマーの増幅効率を検出する必要があります。有効な qRT-PCR 増幅効率は 85% ~ 115% であることが認識されています。次の 2 つの方法があります。
1. 標準曲線法:
a.ミックスcDNA
b.勾配希釈
c.qPCR
d.増幅効率を計算するための線形回帰式
2. リンレグPCR
LinRegPCR は、SYBR Green または同様の化学に基づく定量的 PCR (qPCR) データとも呼ばれる、リアルタイム RT-PCR データを分析するためのプログラムです。このプログラムは、非ベースライン補正データを使用し、各サンプルで個別にベースライン補正を実行し、直線性の範囲を決定し、線形回帰分析を使用して PCR データセット全体に直線を当てはめます。この線の傾きから、個々のサンプルの PCR 効率が計算されます。アンプリコンあたりの平均 PCR 効率とサンプルあたりの Ct 値を使用して、サンプルあたりの開始濃度を計算し、任意の蛍光単位で表します。データの入出力は Excel スプレッドシートを介して行われます。サンプルのみ
混合は必要ですが、勾配はありません
必要な手順は次のとおりです。(Bole CFX96 を例に挙げますが、明確な ABI を備えたマシンとは言えません)
実験:これは標準的な qPCR 実験です。
qPCR データ出力:LinRegPCR は、RDML または定量化増幅結果の 2 つの形式の出力ファイルを認識できます。実際には、サイクル数と蛍光シグナルを機械でリアルタイムに検出した値であり、増幅率は線形セグメント効率の蛍光変化値を解析することで得られます。
データの選択: 理論的には、RDML 値は使用できるはずです。私のパソコンの問題は、ソフトウェアがRDMLを認識できないことだと思われますので、Excelの出力値を元データとして持っています。サンプルの追加に失敗しているなど、最初にデータの大まかなスクリーニングを行うことをお勧めします。出力データ内のポイントは削除できます(もちろん削除できません。LinRegPCR は後の段階でこれらのポイントを無視します)
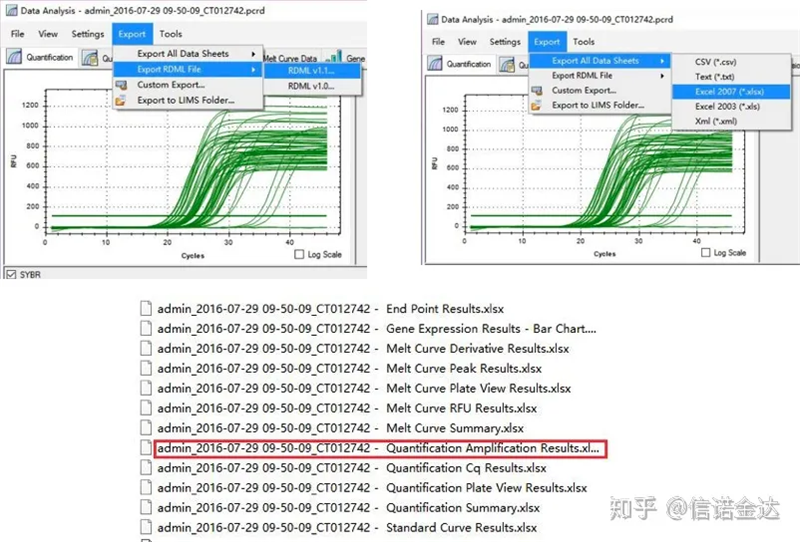
図5 qPCRデータのエクスポート
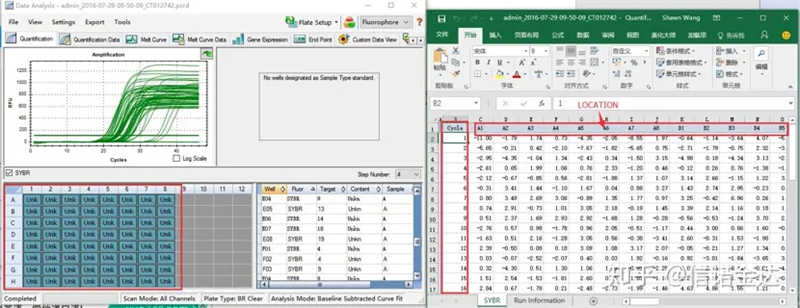
図6 候補サンプルの選定
データ入力:Qualification Amplification results.xls を開く → LinRegPCR を開く → ファイル → Excel から読み取る → 図 7 に示すようにパラメータを選択 → OK → ベースラインの決定をクリック
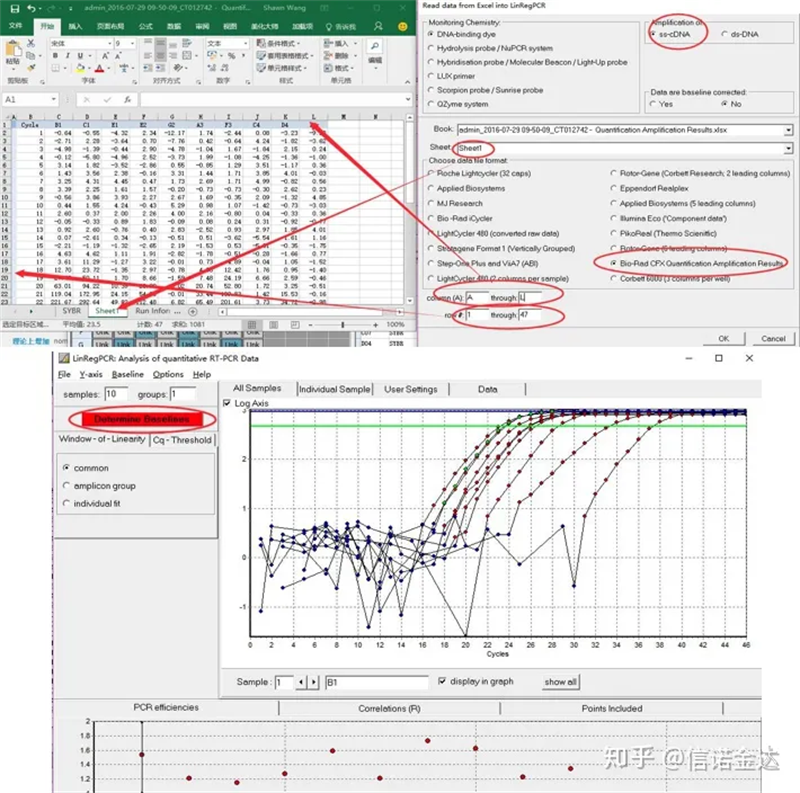
図7 linRegPCRデータ入力のステップ
結果:繰り返しがない場合、グループ化は必要ありません。繰り返しがある場合は、サンプルグループ化でグループ化を編集し、識別子に遺伝子名を入力すると、同じ遺伝子が自動的にグループ化されます。最後に、ファイルをクリックして Excel をエクスポートし、結果を確認します。各ウェルの増幅効率とR2結果が表示されます。次に、グループに分けると補正後の平均増幅効率が表示されます。各プライマーの増幅効率が 85% ~ 115% であることを確認してください。大きすぎたり小さすぎたりすると、プライマーの増幅効率が悪いということになります。
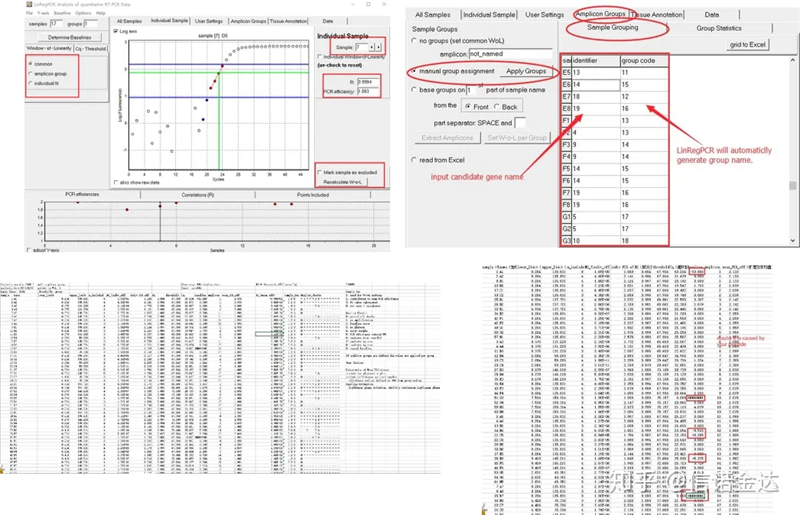
図 8 結果とデータ出力
実験プロセス:
RNA の品質要件:
純度:1.72.0 は、残留イソチオシアネートが存在する可能性があることを示します。きれいな核酸 A260/A230 は 2 程度であるはずです。230 nm に強い吸収がある場合は、フェネート イオンなどの有機化合物が存在することを示します。また、1.5%アガロースゲル電気泳動でも検出できます。ssRNA は変性が無く、分子量の対数が直線関係にないため、分子量を正確に表現できません。集中力: 理論上いいえ100ng/ul未満、濃度が低すぎる場合、一般に純度は高くなく低い
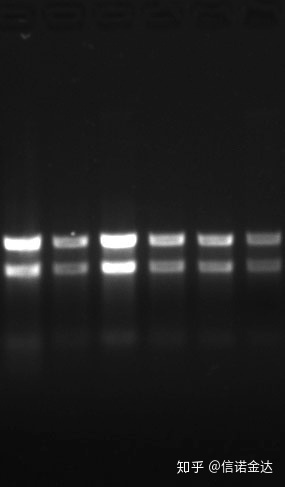
図9 RNAゲル
さらに、サンプルが貴重で RNA 濃度が高い場合は、抽出後に分注し、逆転写のために RNA を最終濃度 100 ~ 300 ng/ul に希釈することをお勧めします。の逆転写のプロセス、mRNA が転写されるとき、polyA テールに特異的に結合できるオリゴ (dt) プライマーが逆転写に使用されますが、lncRNA と circRNA はトータル RNA の逆転写にランダム 6 量体 (ランダム 6 量体) プライマーを使用します。miRNA の場合、miRNA 特異的なネックループ プライマーが逆転写に使用されます。現在、多くの企業が特別な尾引きキットを発売しています。ステムループ法では、テーリング法がより便利で、ハイスループットで、試薬の節約に優れていますが、同じファミリーの miRNA を区別する効果はステムループ法ほど良くないはずです。各逆転写キットには、遺伝子特異的プライマー (ステムループ) の濃度に関する要件があります。miRNA に使用される内部参照は U6 です。ステムループ反転のプロセスでは、U6 のチューブを個別に反転し、U6 の前後のプライマーを直接追加する必要があります。circRNA と lncRNA はどちらも内部参照として HKG を使用できます。のcDNA検出、
RNA が問題なければ、cDNA も問題ありません。ただし、実験の完璧さを追求する場合は、gDNA と cds を区別できる内部参照遺伝子 (リファレンス遺伝子、RG) を使用するのが最善です。一般に、RG はハウスキーピング遺伝子です。、HKG)図10に示すように。当時、私は大豆貯蔵タンパク質を作っており、イントロンを含むアクチン7を内部標準として使用していました。このプライマーの増幅断片のサイズは、gDNAでは452bp、cDNAを鋳型にすると142bpとなる。そして、検査の結果、実際にはcDNAの一部にgDNAが混入していることが判明し、逆転写結果にも問題がなく、PCRの鋳型として使用できることが判明した。cDNA を直接アガロースゲル電気泳動するのは無駄であり、拡散したバンドなので説得力がありません。
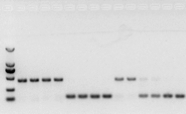
図 10 cDNA の検出
qPCR条件の決定キットのプロトコールに従い、主に tm 値の段階で概ね問題ありません。プライマー設計時に一部のプライマーが適切に設計されておらず、その結果、tm 値と理論上の 60°C の間に大きな差が生じた場合は、サンプルを混合した後、cDNA をプライマーでグラジエント PCR を実行し、バンドのない温度を TM 値として設定しないようにすることをお勧めします。
データ分析
従来の相対蛍光定量的 PCR 処理方法は、基本的に 2 に従っています。-ΔΔCT。データ処理テンプレート。
関連製品:
RT Easy I (ファーストストランド cDNA 合成用マスタープレミックス)
RT Easy II (qPCR 用ファーストストランド cDNA 合成用マスタープレミックス)
投稿日時: 2023 年 3 月 14 日



