



1. 基礎知識(実験編を見たい場合は直接後編へお進みください)
リアルタイムPCRは、従来のPCRの派生反応として、主にPCR増幅反応の各サイクルにおける増幅産物量の変化を蛍光シグナルの変化によってリアルタイムにモニタリングし、ct値と検量線との関係から出発鋳型を定量分析するものです。
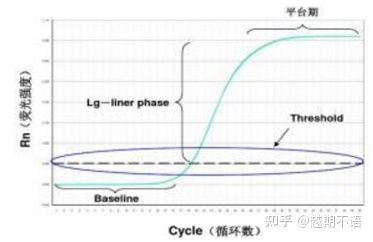
RT-PCRの具体的なデータは以下の通りです。ベースライン, 蛍光閾値とCt値。
| ベースライン: | 3 ~ 15 サイクルの蛍光値がベースライン (ベースライン) ですが、これは測定の時々の誤差によって引き起こされます。 |
| しきい値 (しきい値): | 増幅曲線の指数関数的増加領域の適切な位置に設定された蛍光検出限界を指し、通常はベースラインの標準偏差の 10 倍です。 |
| CT値: | 各反応チューブ内の蛍光値が閾値に達するPCRサイクル数です。 Ct 値は、初期テンプレートの量に反比例します。 |
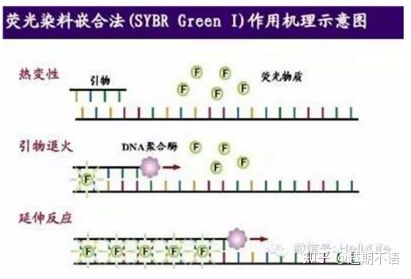
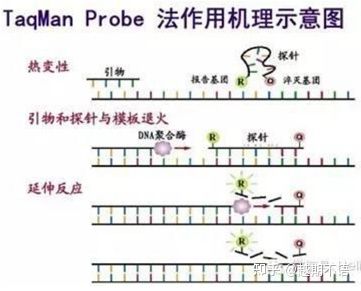
RT-PCR の一般的な標識方法:
| 方法 | アドバンテージ | 欠点がある | 適用範囲 |
| SYBRグリーンⅠ | 適用範囲が広く、感度が高く、安価で便利です | プライマーの要件が高く、非特異的なバンドが発生しやすい | さまざまな対象遺伝子の定量解析、遺伝子発現の研究、遺伝子組換え動植物の研究などに適しています。 |
| タクマン | 優れた特異性と高い再現性 | 価格は高く、特定の目的にのみ適しています。 | 病原体の検出、薬剤耐性遺伝子の研究、薬効評価、遺伝性疾患の診断。 |
| 分子ビーコン | 高い特異性、蛍光、低いバックグラウンド | 価格が高い、特定の用途にしか適さない、デザインが難しい、価格が高い。 | 特異的遺伝子解析、SNP解析 |
2. 実験手順
2.1 実験的なグループ分けについて- グループ内に複数のウェルが存在し、生物学的反復が存在する必要があります。
| ① | ブランクコントロール | 実験で細胞の増殖状態を検出するために使用されます。 |
| ② | ネガティブコントロール siRNA (非特異的 siRNA 配列) | RNAi 作用の特異性を実証します。siRNA は、200nM の濃度で非特異的ストレス応答を誘発する可能性があります。 |
| ③ | トランスフェクション試薬の制御 | トランスフェクション試薬の細胞に対する毒性や標的遺伝子の発現への影響を除外します。 |
| ④ | 標的遺伝子に対するsiRNA | 標的遺伝子の発現をノックダウンする |
| ⑤(オプション) | 陽性siRNA | 実験システムおよび運用上の問題のトラブルシューティングに使用されます |
| ⑥(オプション) | 蛍光コントロールsiRNA | 細胞トランスフェクションの効率は顕微鏡で観察できます |
2.2 プライマー設計の原則
| 増幅された断片のサイズ | 好ましくは100~150bp |
| プライマーの長さ | 18-25bp |
| GC コンテンツ | 30%~70%、できれば45%~55% |
| Tm値 | 58~60℃ |
| 順序 | T/C 連続を避けてください。A/G連続 |
| 3 エンドシーケンス | GC リッチまたは AT リッチは避けてください。末端塩基は、好ましくはGまたはCであり、Tを避けるのが最善です |
| 相補性 | プライマー内または 2 つのプライマー間に 3 塩基を超える相補配列を避けてください。 |
| 特異性 | ブラスト検索を使用してプライマーの特異性を確認する |
①siRNAは種特異的であり、種が異なれば配列も異なります。
②siRNAは凍結乾燥粉末で包装されており、室温で2~4週間安定に保存できます。
2.3 事前に準備が必要な道具や試薬
| プライマー (内部参照) | 前進・後進2本含む |
| プライマー(標的遺伝子) | 前進・後進2本含む |
| ターゲット Si RNA (3 ストリップ) | 通常、同社は 3 つのストリップを合成し、RT-PCR によって 3 つのうちの 1 つを選択します。 |
| トランスフェクションキット | リポ2000など |
| RNA迅速抽出キット | トランスフェクション後のRNA抽出用 |
| 高速逆転写キット | cDNA合成用 |
| PCR増幅キット | 2×スーパーSYBRグリーン qPCRマスターミックス |
2.4 特定の実験手順で注意する必要がある問題について:
①siRNA導入工程
1. プレーティングには、24 ウェル プレート、12 ウェル プレート、または 6 ウェル プレートを選択できます (24 ウェル プレートの各ウェルで提案されている平均 RNA 濃度は約 100 ~ 300 ng/uL)。細胞の最適なトランスフェクション密度は最大 60 % ~ 80% 程度です。
2. トランスフェクションの手順と特定の要件は、指示に厳密に従っています。
3. トランスフェクション後、mRNA 検出 (RT-PCR) またはタンパク質検出 (WB) のために 24 ~ 72 時間以内にサンプルを収集できます。
②RNA抽出工程
1. 外因性酵素による汚染を防ぎます。主にマスクと手袋の厳密な着用が含まれます。滅菌済みのピペットチップとEPチューブを使用します。実験に使用する水はRNaseフリーである必要があります。
2. 迅速抽出キットに記載されているように 2 回行うことをお勧めします。これにより、純度と収率が大幅に向上します。
3. 廃液が RNA カラムに触れないようにしてください。
③RNA定量
RNA を抽出した後、Nanodrop を使用して直接定量することができ、最小読み取り値は 10 ng/ul という低い値になります。
④逆転写工程
1. RT-qPCR は感度が高いため、後続の Ct が異なりすぎたり、SD が統計分析するには大きすぎたりすることを防ぐために、サンプルごとに少なくとも 3 つの平行ウェルを作成する必要があります。
2. マスターミックスの凍結融解を繰り返し行わないでください。
3. 各チューブ/穴は新しいチップと交換する必要があります。サンプルを追加するために同じピペット チップを継続的に使用しないでください。
4. サンプル添加後の 96 ウェルプレートに貼付されたフィルムは、プレートで平滑にする必要があります。チューブの壁についた液体を流して気泡を取り除くために、機械に置く前に遠心分離するのが最善です。
⑤コモンカーブ解析
| 対数成長期がない | おそらく高濃度のテンプレート |
| CT値なし | 蛍光シグナルを検出するための不適切な手順。 プライマーまたはプローブの分解 - その完全性は PAGE 電気泳動によって検出できます。 テンプレートの量が不十分です。 テンプレートの劣化 – 不純物の混入やサンプル調製時の凍結と解凍の繰り返しを回避します。 |
| カット>38 | 増幅効率が低い。PCR 産物が長すぎます。さまざまな反応成分が分解される |
| 直線的な増幅曲線 | プローブは、凍結融解サイクルを繰り返すか、光に長時間さらされると部分的に劣化する可能性があります。 |
| 特に重複ホールの差が大きい | 反応液が完全に溶けていない、または反応液が混合していない。PCR装置の温浴槽が蛍光物質で汚染されている |
2.5 データ分析について
qPCR のデータ解析は、相対定量と絶対定量に分けられます。たとえば、治療グループの細胞を対照グループの細胞と比較した場合、
X 遺伝子の mRNA が何回変化するか、これは相対的な定量化です。特定の数の細胞では、X 遺伝子の mRNA
コピーが何枚あるか、これは絶対的な定量化です。通常、研究室で最もよく使用されるのは相対定量法です。いつもの、2-ΔΔct法は実験で最もよく使われる方法なので、ここではこの方法のみを詳しく紹介します。
2-ΔΔct 法: 得られる結果は、対照群の標的遺伝子に対する実験群の標的遺伝子の発現の差です。標的遺伝子と内部参照遺伝子の両方の増幅効率が 100% に近く、相対偏差が 5% を超えないことが必要です。
計算方法は以下のとおりです。
Δct 対照群 = 対照群の標的遺伝子の ct 値 – 対照群の内部参照遺伝子の ct 値
Δct 実験群 = 実験群の標的遺伝子の ct 値 – 実験群の内部参照遺伝子の ct 値
ΔΔct=Δct 実験グループ - Δct 対照グループ
最後に、発現レベルの差の倍数を計算します。
Change Fold=2-ΔΔct (Excel関数のPOWERに相当)
関連製品:
投稿時刻: 2023 年 5 月 20 日



